Саксенда® инструкция по применению
Состав и форма выпуска
ФАРМАКОЛОГІЧНІ ВЛАСТИВОСТІ:
фармакодинаміка.
Механізм дії. Ліраглутид є ацильованим аналогом ГПП–1 з послідовністю амінокислот на 97% гомологічною ендогенному людському ГПП–1, що зв’язується з ГПП-1-рецепторами та активує їх. ГПП–1 є фізіологічним регулятором апетиту та споживання їжі, але точний механізм його дії повністю не встановлений. У дослідженнях на тваринах периферійне введення ліраглутиду призвело до його накопичення в специфічних ділянках мозку, що відповідають за регуляцію апетиту, де ліраглутид завдяки специфічній активації рецептора ГПП–1Р підвищував відчуття насичення і знижував сигнали голоду, що сприяло зниженню маси тіла.Рецептори ГПП–1 також експресуються у певних ділянках серця, судин, імунній системі та нирках. При моделюванні атеросклерозу у мишей ліраглутид запобігав прогресуванню аортальної бляшки та знижував запалення в бляшці. Крім того, ліраглутид позитивно впливав на ліпіди плазми. Ліраглутид не зменшував розмір вже наявних бляшок.
Фармакодинамічні ефекти. Зниження маси тіла відбувається завдяки переважній втраті вісцерального жиру порівняно з підшкірним. Ліраглутид регулює апетит, підсилюючи відчуття ситості та наповненості шлунка, знижуючи при цьому відчуття голоду, та призводить до зниження споживання їжі. Ліраглутид не збільшує енерговитрати порівняно з плацебо. Ліраглутид стимулює секрецію інсуліну та зменшує надмірно високу секрецію глюкагону залежно від рівня глюкози, що призводить до зниження глюкози натще та після вживання їжі.У пацієнтів з переддіабетом та цукровим діабетом ефект зниження рівня глюкози більш виражений, порівняно з пацієнтами з нормоглікемією. Клінічні випробування свідчать, що ліраглутид покращує та підтримує функцію бета-клітин відповідно до НОМА-В та співвідношення проінсулін/інсулін.
Клінічна ефективність та безпека. Клінічна ефективність та безпека ліраглутиду при застосуванні для зниження маси тіла як доповнення до дієти зі зниженою калорійністю та збільшеною фізичною активністю були вивчені у чотирьох рандомізованих подвійних сліпих плацебо-контрольованих дослідженнях фази 3 за участі 5358 пацієнтів.
Дослідження 1 (SCALE Ожиріння та переддіабет — 1839): Всього 3731 пацієнт із ожирінням (індекс маси тіла (ІМТ) ≥30 кг/м2) або з надмірною масою тіла (ІМТ ≥27 кг/м2), який страждає на дисліпідемію та/або гіпертензію, були стратифіковані за допомогою скринінгу відповідно до статусу переддіабету та початкового рівня ІМТ (≥30 кг/м2 або <30 кг/м2). Всі пацієнти (3731) були рандомізовані за тривалістю лікування 56 тижнів, а 2254 пацієнти, які страждали на переддіабет, при скринінгу були рандомізовані на 160 тижнів лікування. Обидва періоди лікування супроводжувались 12-тижневим періодом спостереження за групами препарат/плацебо. Корекція способу життя у вигляді дієти зі зниженою калорійністю та збільшеною фізичною активністю була базовою терапією для всіх пацієнтів.56-тижневе дослідження 1 показало втрату маси тіла у всіх (3731) рандомізованих пацієнтів (2590 пацієнтів завершили курс лікування). 160-тижневе дослідження 1 показало час до розвитку цукрового діабету 2 типу у 2254 рандомізованих пацієнтів, хворих на переддіабет (1128 пацієнтів завершили курс лікування).
Дослідження 2 (SCALE Діабет — 1922):Дослідження тривалістю 56 тижнів, у якому оцінювали втрату маси тіла у 846 рандомізованих пацієнтів із ожирінням та надмірною масою тіла (628 пацієнтів завершили курс лікування), які мали недостатньо контрольований цукровий діабет 2 типу (HbA1c в діапазоні 7–10%). Основним методом лікування на початку дослідження була або дієта, або збільшена фізична активність, або застосування окремих лікарських засобів, таких як метформін, сульфонілсечовина та глітазон, або їх комбінацій.
Дослідження 3 (SCALE Апное під час сну — 3970): Дослідження тривалістю 32 тижні, в якому оцінювали ступінь тяжкості апное під час сну та зниження маси тіла у 359 рандомізованих пацієнтів (276 пацієнтів завершили курс лікування), які страждали на ожиріння та середній або тяжкий ступінь обструктивного апное під час сну.
Дослідження 4 (SCALE Підтримуюче лікування — 1923): Дослідження тривалістю 56 тижнів, в якому оцінювали підтримуюче лікування після втрати маси тіла ≥5%, спричиненої дієтою зі зниженою калорійністю у 422 рандомізованих пацієнтів із ожирінням та надмірною масою тіла (305 пацієнтів завершили курс лікування), які страждали на гіпертензію чи дисліпідемію.
Маса тіла. При застосуванні ліраглутиду було досягнуто зниження маси тіла порівняно з плацебо у пацієнтів із ожирінням та надмірною масою тіла у всіх досліджуваних групах. У більшості пацієнтів втрата маси тіла досягала ≥5% та >10% при застосуванні ліраглутиду порівняно з плацебо. Протягом 160 тижнів дослідження 1 переважна втрата маси тіла відбулася в перший рік терапії і тривала протягом всіх 160 тижнів.• У дослідженні 1 середня втрата маси тіла на 56-му тижні становила 8,0% (8,4 кг) при застосуванні ліраглутиду та 2,6% (2,8 кг) при застосуванні плацебо (розрахункова різниця при лікуванні (РРЛ) (середня втрата у процентах): -5,4 [95% ДІ -5,8; -5,0], р<0,0001, РРЛ (середня втрата у кілограмах)): -5,6 [95% ДІ -6,0; -5,1], p<0,0001). Частина пацієнтів, які втратили 5% та 10% маси тіла на 56-му тижні, становила 63,5% та 32,8% відповідно при застосуванні ліраглутиду проти 26,6 % та 10,1% відповідно при застосуванні плацебо (коефіцієнт вірогідності (втрати ≥5% маси тіла): 4,8 [95% ДІ 4,1; 5,6], р<0,0001, коефіцієнт вірогідності (втрати >10% маси тіла): 4,3 [95 % ДІ 3,5; 5,3] р<0,0001).• У дослідженні 1 середня втрата маси тіла на 160-му тижні становила 6,2% (6,5 кг) при застосуванні ліраглутиду та 1,8% (2,0 кг) при застосуванні плацебо РРЛ (середня втрата у процентах): -4,3 [95 % ДІ -4,9; -3,7], р<0,0001), РРЛ (середня втрата у кілограмах): -4,6 [95% ДІ -5,3; -3,9], p<0,0001). Частина пацієнтів, які втратили 5% та 10% маси тіла на 160-му тижні, становила 49,6% та 24,4% відповідно при застосуванні ліраглутиду проти 23,4 % та 9,5 % відповідно при застосуванні плацебо (коефіцієнт вірогідності (втрати ≥5 % маси тіла): 3,2 [95% ДІ 2,6; 3,9], р<0,0001, коефіцієнт вірогідності (втрати >10% маси тіла): 3,1 [95% ДІ 2,3; 4,1] р<0,0001).• У дослідженні 2 середня втрата маси тіла на 56-му тижні становила 5,9% (6,2 кг) при застосуванні ліраглутиду та 2,0% (2,2 кг) при застосуванні плацебо РРЛ (середня втрата у процентах): -4,0 [95% ДІ -4,8; -3,1], р<0,0001), РРЛ (середня втрата у кілограмах): -4,1 [95% ДІ -5,0; -3,1], p<0,0001). Частина пацієнтів, які втратили 5% та 10% маси тіла на 56-му тижні, становила 49,8% та 22,9% відповідно при застосуванні ліраглутиду проти 13,5% та 4,2% відповідно при застосуванні плацебо (коефіцієнт вірогідності (втрати ≥5% маси тіла): 6,4 [95% ДІ 4,1; 10,0], р<0,0001, коефіцієнт вірогідності (втрати >10% маси тіла): 6,8 [95% ДІ 3,4; 13,8] р<0,0001).• У дослідженні 3 середня втрата маси тіла на 32-му тижні становила 5,7% (6,8 кг) при застосуванні ліраглутиду та 1,6% (1,8 кг) при застосуванні плацебо РРЛ (середня втрата у процентах): -4,2 [95% ДІ -5,2; -3,1], р<0,0001), РРЛ (середня втрата у кілограмах): -4,9 [95% ДІ -6,2; -3,7], p<0,0001). Частина пацієнтів, які втратили 5% маси тіла на 32-му тижні, становила 46,4% при застосуванні ліраглутиду проти 18,1% відповідно при застосуванні плацебо (оцінене співвідношення шансів: 3,9 [95% ДІ 2,4; 6,4], р<0,0001).• У дослідженні 4 більшість пацієнтів підтримували масу тіла, досягнуту на початку лікування ліраглутидом порівняно з плацебо (81,4% та 48,9% відповідно). Середня втрата маси тіла на 56-му тижні становила 6,3% (6,0 кг) при застосуванні ліраглутиду та 0,2% (0,2 кг) при застосуванні плацебо (РРЛ (середня втрата у процентах): -6,1 [95% ДІ -7,5; -4,6], р<0,0001), РРЛ (середня втрата у кілограмах): -5,9 [95% ДІ -7,3; -4,4], p<0,0001). Частина пацієнтів, які втратили 5% та 10% маси тіла на 56-му тижні, становила 50,7% та 27,4% відповідно при застосуванні ліраглутиду проти 21,3% та 6,8% відповідно при застосуванні плацебо (коефіцієнт вірогідності (втрати ≥5% маси тіла): 3,8 [95% ДІ 2,4; 6,0], р<0,0001, коефіцієнт вірогідності (втрати >10% маси тіла): 5,1 [95% ДІ 2,7; 9,7] р<0,0001). Дані щодо втрати маси тіла, терміну лікування та кумулятивного розподілу зміни маси тіла (%) представлені на рисунках 1, 2 і 3.
Втрата маси тіла через 12 тижнів при лікуванні ліраглутидом (3,0 мг). Після 12 тижнів застосування ліраглутиду зареєстровано втрату маси тіла ≥5% (4 тижні — ескалація дози та 12 тижнів — лікувальна доза) у 67,5% пацієнтів у дослідженні 1 тривалістю 56 тижнів. У дослідженні 2 50,4% пацієнтів досягли втрати маси тіла ≥5%,через 12 тижнів. Після застосування ліраглутиду протягом року у 86,2% пацієнтів зниження маси тіла становило ≥5%, а у 51% пацієнтів — ≥10%. Середня втрата маси тіла у пацієнтів, які застосовували ліраглутид протягом року, становила 11,2% від їх початкової маси тіла (9,7% для чоловіків і 11,6% для жінок). Частка пацієнтів, які досягли втрати маси тіла після 12 тижнів терапії <5% та які після року застосування ліраглутиду не досягли втрати маси тіла ≥10%, становила 93,4%.
Контроль глікемії. Лікування ліраглутидом значно покращило показники глікемії у пацієнтів з нормоглікемією, переддіабетом та цукровим діабетом 2 типу. У 56-тижневій частині дослідження 1 цукровий діабет 2 типу розвинувся у меншої кількості пацієнтів, які отримували ліраглутид, порівняно з пацієнтами, які отримували плацебо (0,2% проти 1,1% відповідно). У більшості пацієнтів з переддіабетом на початку лікування спостерігався зворотний розвиток даного захворювання після застосування ліраглутиду порівняно з плацебо (69,2% проти 32,7% відповідно). Порівнюючи з початковим значенням HbA1c, у 5,6% пацієнтів, які застосовували ліраглутид, спостерігалося зниження середнього значення HbA1c на 56-му тижні на -0,3% проти -0,1 % пацієнтів, які застосовували плацебо (РРЛ: -0,23 [95% ДІ -0,25; -0,21], р<0,0001). Порівнюючи з початковим рівнем глюкози в плазмі натще (ГПН) 5,3 ммоль/л у пацієнтів, які застосовували ліраглутид, спостерігалось зниження ГПН в середньому на -0,4 ммоль/л проти -0,01 ммоль/л у пацієнтів, які застосовували плацебо на 56-му тижні (РРЛ: -0,38 [95% ДІ -0,42; -0,35], р<0,0001).Показником первинної ефективності 160-тижневої частини дослідження 1 було відношення кількості пацієнтів, у яких виник цукровий діабет 2 типу, до періоду проявлення. На 160-му тижні дослідження у 3% пацієнтів, які отримували лікарський засіб Саксенда®, і в 11% пацієнтів, які отримували плацебо, діагностували цукровий діабет 2 типу. Приблизний період розвитку цукрового діабету 2 типу у пацієнтів, які отримували ліраглутид 3 мг, був у 2,7 раза довший (95% ДІ [1,9; 3,9]), а коефіцієнт ризику розвитку цукрового діабету 2 типу становив 0,2 для ліраглутиду порівняно з плацебо. Порівнюючи з початковим значенням HbA1c, у 5,8% пацієнтів, які застосовували ліраглутид, та у 5,7% пацієнтів, які застосовували плацебо, спостерігалося зниження значення HbA1c на 160-му тижні в середньому на 0,4% та 0,1% відповідно (РРЛ: -0,21 [95% ДІ -0,24; -0,18], р<0,0001). Порівнюючи з початковим значенням ГПН (5,5 ммоль/л), у пацієнтів, які застосовували ліраглутид, спостерігалось зниження ГПН в середньому на 0,4 ммоль/л проти 0,04 ммоль/л у пацієнтів, які застосовували плацебо на 160-му тижні (РРЛ: -0,4 [95% ДІ -0,5; -0,4], р<0,0001). У дослідженні 2, порівнюючи з початковим значенням HbA1c, у 7,9% пацієнтів, які застосовували ліраглутид, спостерігалося зниження середнього значення HbA1c на 56-му тижні на 1,3 % проти 0,4% пацієнтів, які застосовували плацебо (РРЛ: -0,9 [95% ДІ -1,1; -0,8], р<0,0001). Порівнюючи з початковим значенням ГПН 8,8 ммоль/л для пацієнтів, які застосовували ліраглутид, та 8,6 ммоль/л для пацієнтів, які застосовували плацебо, у пацієнтів, які застосовували ліраглутид, спостерігалось зниження ГПН в середньому на 1,9 ммоль/л проти 0,1 ммоль/л у пацієнтів, які застосовували плацебо на 56-му тижні (РРЛ: -1,8 [95% ДІ -2,1; -1,4], р<0,0001).
Кардіометаболічні фактори ризику. Лікування ліраглутидом значно покращило показники систолічного артеріального тиску, також спостерігалось зменшення окружності талії порівняно з плацебо. У дослідженні 1 початкове значення систолічного артеріального тиску становило 123,00 мм рт. ст., на 56-му тижні спостерігалося зниження систолічного артеріального тиску в середньому на 4,3 мм рт. ст., та на 1,5 мм рт. ст. у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -2,8 [95% ДІ -3,6; -2,1], р<0,0001). Початкове значення діастолічного артеріального тиску становило 78,7 мм рт. ст. у пацієнтів, які застосовували ліраглутид, та 78,9 мм рт. ст. у пацієнтів, які застосовували плацебо; на 56-му тижні спостерігалося зниження діастолічного артеріального тиску на -2,7 мм рт. ст. та 1,8 мм рт. ст. у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -0,9 [95% ДІ -1,4; -0,4], р<0,05). Початкове значення окружності талії становило 115,0 см для пацієнтів, які застосовували ліраглутид, та 114,5 см для пацієнтів, які застосовували плацебо; на 56-му тижні спостерігалося зменшення окружності талії на -8,2 см та на -4,0 см у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -4,2 [95% ДІ -4,7; -3,7], р<0,0001). У дослідженні 1 початкове значення систолічного артеріального тиску становило 124,8 мм рт. ст. для пацієнтів, які застосовували ліраглутид, та 125,0 мм рт. ст. для пацієнтів, які застосовували плацебо; на 160-му тижні спостерігалося зниження систолічного артеріального тиску на -3,2 мм рт. ст., та на -0,4 мм рт. ст. у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -2,8 [95% ДІ -3,8; -1,8], р<0,0001). Початкове значення діастолічного артеріального тиску становило 79,4 мм рт. ст. для пацієнтів, які застосовували ліраглутид, та 79,8 мм рт. ст. для пацієнтів, які застосовували плацебо; на 160-му тижні спостерігалось зниження діастолічного артеріального тиску на 2,4 мм рт. ст. та -1,7 мм рт. ст. у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -0,6 [95% ДІ -1,3; -0,1]). Початкове значення окружності талії становило 116,6 см для пацієнтів, які застосовували ліраглутид, та 116,7 см для пацієнтів, які застосовували плацебо; на 160-му тижні спостерігалось зменшення окружності талії на 6,9 см та на 3,4 см у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -3,5 [95% ДІ -4,2; -2,8], р<0,0001). У дослідженні 2 початкове значення систолічного артеріального тиску становило 128,9 мм рт. ст. у пацієнтів, які застосовували ліраглутид, та 129,2 мм рт. ст. у пацієнтів, які застосовували плацебо; на 56-му тижні спостерігалось зниження систолічного артеріального тиску на 3,0 мм рт. ст. та на 0,4 мм рт. ст. у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -2,6 [95% ДІ -4,6; -0,6], р<0,0001). Початкове значення діастолічного артеріального тиску становило 79 мм рт. ст. для пацієнтів, які застосовували ліраглутид, та 79,3 мм рт. ст. для пацієнтів, які застосовували плацебо; на 56-му тижні спостерігалось зниження діастолічного артеріального тиску на 1,0 мм рт. ст. та 0,6 мм рт. ст. у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -0,4 [95% ДІ -1,7; -1,0], р=0,5918). Початкове значення окружності талії становило 118,1 см для пацієнтів, які застосовували ліраглутид, та 117,3 см для пацієнтів, які застосовували плацебо; на 56-му тижні спостерігалось зменшення окружності талії на 6,0 см та на 2,8 см у пацієнтів, які застосовували ліраглутид та плацебо відповідно (РРЛ: -3,2 [95% ДІ -4,2; -2,2], р<0,0001).
Індекс апное-гіпное (ІАГ). При застосуванні ліраглутиду спостерігалось значне зниження тяжкості обструктивного апное під час сну порівняно з плацебо, яке було оцінене за допомогою зменшення порівняно з плацебо (12,2 події/година для ліраглутиду проти 6,1 події/година для плацебо (РРЛ: -6,1 [95% ДІ -11,0; -1,2], р<0,05).
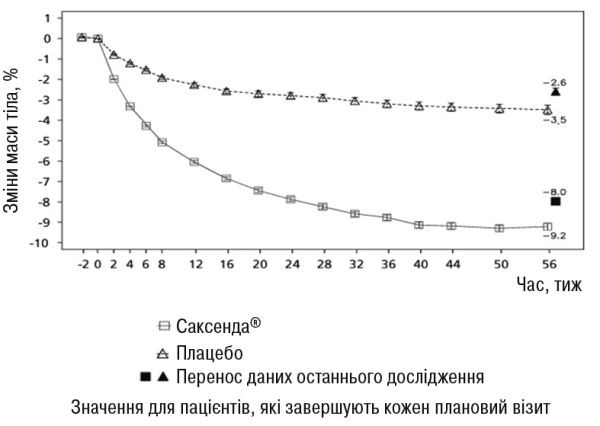 Рисунок 1. Зміна маси тіла (%) від початкового показника за часом у дослідженні 1 (0–56 тижнів)
Рисунок 1. Зміна маси тіла (%) від початкового показника за часом у дослідженні 1 (0–56 тижнів)
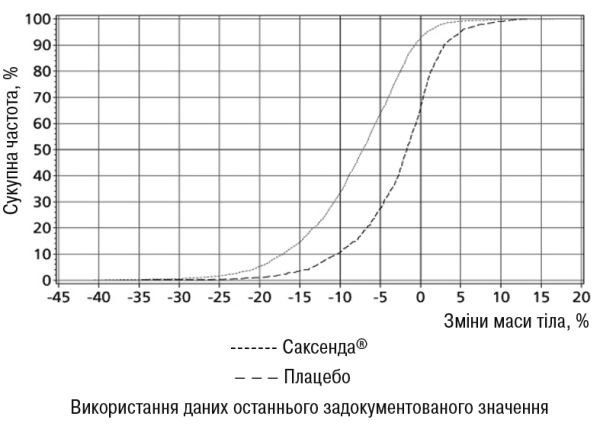 Рисунок 2. Кумулятивний розподіл зміни маси тіла (%) після 56 тижнів лікування у дослідженні 1
Рисунок 2. Кумулятивний розподіл зміни маси тіла (%) після 56 тижнів лікування у дослідженні 1
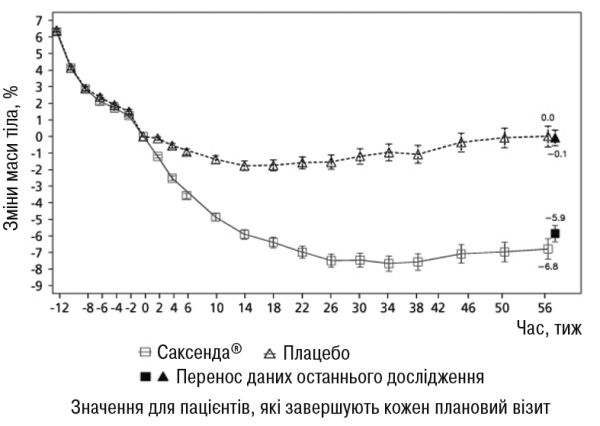 Рисунок 3. Зміна рандомізації (тиждень 0) в масі тіла (%) за часом у дослідженні 4До тижня 0 пацієнти знаходилися лише на дієті зі зниженою калорійністю, також для них було збільшено фізичну активність. На тижні 0 пацієнти рандомізовано отримували або лікарський засіб Саксенда®, або плацебо.
Імуногенність. Враховуючи потенційні імуногенні властивості лікарських засобів, що містять білки або пептиди, можна передбачити, що в пацієнтів можуть утворитися антитіла до ліраглутиду після застосування лікарського засобу Саксенда®. Під час клінічних досліджень у 2,5% пацієнтів, які отримували ліраглутид, утворювались антитіла до ліраглутиду. Утворення антитіл не призводило до зниження ефективності ліраглутиду.
Вплив на серцево-судинну систему. Основні серйозні несприятливі серцево-судинні явища (МАСЕ), які були визначені зовнішньою незалежною експертною групою: нелетальний інфаркт міокарда, нелетальний інсульт, летальний випадок через серцево-судинну патологію. В усіх тривалих клінічних дослідженнях лікарського засобу Саксенда® було зареєстровано 6 МАСЕ у пацієнтів, які отримували ліраглутид, та 10 МАСЕ у пацієнтів, які отримували плацебо. Відношення ризиків та ДІ 95% при порівнянні лікарського засобу Саксенда® та плацебо становило 0,33 [0,12; 0,90].У клінічних дослідженнях 3-ї фази спостерігалось незначне підвищення частоти серцевих скорочень на 2,5 удара за хвилину (від 1,6 до 3,6 удара за хвилину в окремих дослідженнях). Максимальне збільшення частоти серцевих скорочень спостерігалося приблизно після 6 тижнів терапії. Тривалий клінічний вплив збільшення частоти серцевих скорочень не встановлений. Це збільшення було зворотним та зникало після припинення терапії ліраглутидом (див. ОСОБЛИВОСТІ ЗАСТОСУВАННЯ).У дослідженні LEADER брали участь 9340 пацієнтів із недостатньо контрольованим цукровим діабетом 2 типу. Переважна кількість із них страждали на серцево-судинні захворювання. Пацієнтів рандомізовано розподіляли для застосування ліраглутиду у добовій дозі до 1,8 мг (4668) та плацебо (4672) щодо надання стандартної медичної допомоги в обох групах рандомізації. Тривалість терапії становила від 3,5 до 5 років. Середній вік пацієнтів становив 64 роки, середній ІМТ — 32,5 кг/м2. Середнє значення початкового рівня HbA1c становило 8,7 і покращилося через 3 роки на 1,2% у пацієнтів, яким був призначений ліраглутид, та на 0,8% у пацієнтів, яким було призначено плацебо. Первинною кінцевою точкою ефективності був час від рандомізації до першого виникнення будь-яких основних MACE: нелетального інфаркту міокарда, нелетального інсульту, летального випадку через серцево-судинну патологію. Ліраглутид значно знизив частоту виникнення основних несприятливих серцево-судинних явищ (події первинної кінцевої точки, MACE) порівняно з плацебо (3,41 проти 3,90 на 100 пацієнто-років у групах ліраглутиду та плацебо відповідно), зменшивши ризик на 13%, HR 0,87, [0,78; 0,97] [95% ДІ]) (р = 0,005) (див. Рисунок 4).
Рисунок 3. Зміна рандомізації (тиждень 0) в масі тіла (%) за часом у дослідженні 4До тижня 0 пацієнти знаходилися лише на дієті зі зниженою калорійністю, також для них було збільшено фізичну активність. На тижні 0 пацієнти рандомізовано отримували або лікарський засіб Саксенда®, або плацебо.
Імуногенність. Враховуючи потенційні імуногенні властивості лікарських засобів, що містять білки або пептиди, можна передбачити, що в пацієнтів можуть утворитися антитіла до ліраглутиду після застосування лікарського засобу Саксенда®. Під час клінічних досліджень у 2,5% пацієнтів, які отримували ліраглутид, утворювались антитіла до ліраглутиду. Утворення антитіл не призводило до зниження ефективності ліраглутиду.
Вплив на серцево-судинну систему. Основні серйозні несприятливі серцево-судинні явища (МАСЕ), які були визначені зовнішньою незалежною експертною групою: нелетальний інфаркт міокарда, нелетальний інсульт, летальний випадок через серцево-судинну патологію. В усіх тривалих клінічних дослідженнях лікарського засобу Саксенда® було зареєстровано 6 МАСЕ у пацієнтів, які отримували ліраглутид, та 10 МАСЕ у пацієнтів, які отримували плацебо. Відношення ризиків та ДІ 95% при порівнянні лікарського засобу Саксенда® та плацебо становило 0,33 [0,12; 0,90].У клінічних дослідженнях 3-ї фази спостерігалось незначне підвищення частоти серцевих скорочень на 2,5 удара за хвилину (від 1,6 до 3,6 удара за хвилину в окремих дослідженнях). Максимальне збільшення частоти серцевих скорочень спостерігалося приблизно після 6 тижнів терапії. Тривалий клінічний вплив збільшення частоти серцевих скорочень не встановлений. Це збільшення було зворотним та зникало після припинення терапії ліраглутидом (див. ОСОБЛИВОСТІ ЗАСТОСУВАННЯ).У дослідженні LEADER брали участь 9340 пацієнтів із недостатньо контрольованим цукровим діабетом 2 типу. Переважна кількість із них страждали на серцево-судинні захворювання. Пацієнтів рандомізовано розподіляли для застосування ліраглутиду у добовій дозі до 1,8 мг (4668) та плацебо (4672) щодо надання стандартної медичної допомоги в обох групах рандомізації. Тривалість терапії становила від 3,5 до 5 років. Середній вік пацієнтів становив 64 роки, середній ІМТ — 32,5 кг/м2. Середнє значення початкового рівня HbA1c становило 8,7 і покращилося через 3 роки на 1,2% у пацієнтів, яким був призначений ліраглутид, та на 0,8% у пацієнтів, яким було призначено плацебо. Первинною кінцевою точкою ефективності був час від рандомізації до першого виникнення будь-яких основних MACE: нелетального інфаркту міокарда, нелетального інсульту, летального випадку через серцево-судинну патологію. Ліраглутид значно знизив частоту виникнення основних несприятливих серцево-судинних явищ (події первинної кінцевої точки, MACE) порівняно з плацебо (3,41 проти 3,90 на 100 пацієнто-років у групах ліраглутиду та плацебо відповідно), зменшивши ризик на 13%, HR 0,87, [0,78; 0,97] [95% ДІ]) (р = 0,005) (див. Рисунок 4).
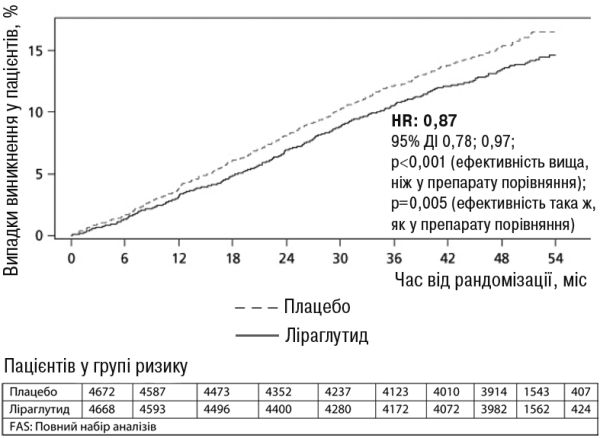 Рисунок 4. Графік часу Каплана-Майєра на початку дослідження MACE – FAS популяції
Фармакокінетика.
Абсорбція Абсорбція ліраглутиду після підшкірного введення відбувається повільно, максимальна концентрація досягається через 11 годин після введення. У пацієнтів, які страждають на ожиріння (ІМТ 30-40 кг/м2), після введення 3 мг ліраглутиду його середня рівноважна концентрація (AUCt/24) досягала приблизно 31 нмоль/л. Експозиція ліраглутиду збільшувалася пропорційно дозі. Абсолютна біодоступність ліраглутиду після підшкірного введення становить приблизно 55%.
Розподіл. Середній видимий об’єм розподілу після підшкірного введення становить 20–25 л (для людини з масою тіла приблизно 100 кг). Ліраглутид зв’язується переважно з білками плазми крові (>98%).
Метаболізм. Протягом 24 годин після введення разової дози [3H]-ліраглутиду здоровим добровольцям основним компонентом у плазмі крові був незмінений ліраглутид. У плазмі крові були виявлені в незначній кількості два метаболіти (≤9% і ≤5% від загального рівня радіоактивності у плазмі крові).
Виведення. Ліраглутид ендогенно метаболізується, як і всі великі білки, без участі специфічного органу як основного шляху елімінації. Після введення дози [3H]-ліраглутиду в сечі і калі не було виявлено незміненого ліраглутиду. Тільки невелика частка введеної радіоактивності у вигляді метаболітів ліраглутиду виводилась нирками та через кишечник (6% і 5% відповідно). Радіоактивні речовини виводяться нирками або через кишечник в основному протягом перших 6 — 8 діб у вигляді трьох метаболітів. Після одноразового підшкірного введення ліраглутиду середнє значення кліренсу становить приблизно 0,9-1,4 л/годину, період напіввиведення — приблизно 13 годин.
Особливі групи пацієнтів
Пацієнти літнього віку. На підставі даних фармакокінетичного аналізу групи пацієнтів віком від 18 до 82 років з надмірною масою тіла чи ожирінням був зроблений висновок, що вік не має клінічно значущого впливу на фармакокінетику ліраглутиду. Тому немає необхідності в коригуванні дози відносно віку.
Стать. Дані фармакокінетичного аналізу показали, що у жінок спостерігається на 24 % нижчий кліренс ліраглутиду порівняно з чоловіками. На підставі цих даних можна зробити висновок, що корекція дози відносно статі не потрібна.
Етнічне походження. На підставі даних фармакокінетичного аналізу групи пацієнтів європеоїдної, монголоїдної, латиноамериканської і негроїдної рас з надмірною масою тіла чи ожирінням був зроблений висновок, що етнічне походження не виявляє будь-якого істотного клінічного впливу на фармакокінетику ліраглутиду.
Маса тіла. Експозиція ліраглутиду зменшується зі збільшенням початкової маси тіла. Як показали дослідження, добова доза ліраглутиду 3,0 мг забезпечує нормальний системний вплив на організм пацієнта з масою тіла 60-234 кг. Експозиція ліраглутиду у пацієнтів з масою тіла більше 234 кг не вивчалась.
Порушення функції печінки. Фармакокінетику ліраглутиду досліджували у пацієнтів із різним ступенем порушень функції печінки у процесі дослідження із застосуванням одноразової дози (0,75 мг). Було показано, що у пацієнтів з легкими і помірними порушеннями функції печінки експозиція ліраглутиду знижувалася на 13-23% порівняно зі здоровими добровольцями. У пацієнтів з тяжкими порушеннями функції печінки (>9 балів за класифікацією Чайлда — П’ю) експозиція була значно нижча (на 44%).
Порушення функції нирок. Експозиція ліраглутиду була знижена у пацієнтів з порушеннями функції нирок порівняно з особами з нормальною функцією нирок у процесі дослідження із застосуванням одноразової дози (0,75 мг). У пацієнтів з легкими порушеннями (кліренс креатиніну 50–80 мл/хв) експозиція знижувалася на 33%, з порушеннями помірної тяжкості (кліренс креатиніну 30–50 мл/хв) — на 14%, з тяжкими порушеннями (кліренс креатиніну <30 мл/хв) — на 27%, а на кінцевих стадіях захворювань нирок, що вимагають проведення діалізу, — на 26%.
Діти. У клінічних дослідженнях фармакокінетичних властивостей брали участь пацієнти з ожирінням віком 12–17 років (14 пацієнтів з масою тіла 80-122 кг) та 7-11 років (16 пацієнтів з масою тіла 45-87 кг). Експозиція ліраглутиду у підлітків (віком 12–17 років) була подібною до експозиції у дорослих з надмірною масою тіла. Порівнювалась експозиція при введенні 3,0 мг ліраглутиду у дорослих, підлітків та дітей віком 7-11 років після корекції маси тіла.
Доклінічні дані з безпеки. Доклінічні дані, що базуються на дослідженнях з фармакологічної безпеки, токсичності повторних доз та генотоксичності, не виявили жодного ризику для людини. У процесі дворічних досліджень канцерогенності у щурів та мишей були виявлені пухлини С-клітин щитовидної залози, що не призводили до летального результату. Нетоксична доза (NOAEL) у щурів не була встановлена. У мавп, що отримували лікування протягом 20 місяців, таких пухлин не виявлено. Пухлини у гризунів обумовлені негенотоксичним специфічним ГПП–1–рецептор-опосередкованим механізмом, до якого частково чутливі гризуни. Значущість цього механізму у людей достатньо низька, але не може бути повністю виключена. Розвитку інших пухлин не було виявлено. У процесі експериментів на тваринах не було виявлено прямого шкідливого впливу на фертильність, проте при введенні найвищих доз відзначалося незначне підвищення ранньої ембріональної летальності. Введення ліраглутиду в період середини вагітності спричиняло зниження маси тіла самки, уповільнення росту плода з нез’ясованим впливом на розвиток ребер у щурів і скелета у кроликів. При введенні ліраглутиду відзначено уповільнення росту новонароджених щурів, що зберігається в період відлучення від годування молоком у групі прийому високої дози. Невідомо, чи уповільнення росту новонароджених щурів обумовлене зниженням споживання ними молока в результаті прямого впливу ГПП–1, чи зменшенням молока у матері, що обумовлено зниженням калорійності споживаної їжі.
Рисунок 4. Графік часу Каплана-Майєра на початку дослідження MACE – FAS популяції
Фармакокінетика.
Абсорбція Абсорбція ліраглутиду після підшкірного введення відбувається повільно, максимальна концентрація досягається через 11 годин після введення. У пацієнтів, які страждають на ожиріння (ІМТ 30-40 кг/м2), після введення 3 мг ліраглутиду його середня рівноважна концентрація (AUCt/24) досягала приблизно 31 нмоль/л. Експозиція ліраглутиду збільшувалася пропорційно дозі. Абсолютна біодоступність ліраглутиду після підшкірного введення становить приблизно 55%.
Розподіл. Середній видимий об’єм розподілу після підшкірного введення становить 20–25 л (для людини з масою тіла приблизно 100 кг). Ліраглутид зв’язується переважно з білками плазми крові (>98%).
Метаболізм. Протягом 24 годин після введення разової дози [3H]-ліраглутиду здоровим добровольцям основним компонентом у плазмі крові був незмінений ліраглутид. У плазмі крові були виявлені в незначній кількості два метаболіти (≤9% і ≤5% від загального рівня радіоактивності у плазмі крові).
Виведення. Ліраглутид ендогенно метаболізується, як і всі великі білки, без участі специфічного органу як основного шляху елімінації. Після введення дози [3H]-ліраглутиду в сечі і калі не було виявлено незміненого ліраглутиду. Тільки невелика частка введеної радіоактивності у вигляді метаболітів ліраглутиду виводилась нирками та через кишечник (6% і 5% відповідно). Радіоактивні речовини виводяться нирками або через кишечник в основному протягом перших 6 — 8 діб у вигляді трьох метаболітів. Після одноразового підшкірного введення ліраглутиду середнє значення кліренсу становить приблизно 0,9-1,4 л/годину, період напіввиведення — приблизно 13 годин.
Особливі групи пацієнтів
Пацієнти літнього віку. На підставі даних фармакокінетичного аналізу групи пацієнтів віком від 18 до 82 років з надмірною масою тіла чи ожирінням був зроблений висновок, що вік не має клінічно значущого впливу на фармакокінетику ліраглутиду. Тому немає необхідності в коригуванні дози відносно віку.
Стать. Дані фармакокінетичного аналізу показали, що у жінок спостерігається на 24 % нижчий кліренс ліраглутиду порівняно з чоловіками. На підставі цих даних можна зробити висновок, що корекція дози відносно статі не потрібна.
Етнічне походження. На підставі даних фармакокінетичного аналізу групи пацієнтів європеоїдної, монголоїдної, латиноамериканської і негроїдної рас з надмірною масою тіла чи ожирінням був зроблений висновок, що етнічне походження не виявляє будь-якого істотного клінічного впливу на фармакокінетику ліраглутиду.
Маса тіла. Експозиція ліраглутиду зменшується зі збільшенням початкової маси тіла. Як показали дослідження, добова доза ліраглутиду 3,0 мг забезпечує нормальний системний вплив на організм пацієнта з масою тіла 60-234 кг. Експозиція ліраглутиду у пацієнтів з масою тіла більше 234 кг не вивчалась.
Порушення функції печінки. Фармакокінетику ліраглутиду досліджували у пацієнтів із різним ступенем порушень функції печінки у процесі дослідження із застосуванням одноразової дози (0,75 мг). Було показано, що у пацієнтів з легкими і помірними порушеннями функції печінки експозиція ліраглутиду знижувалася на 13-23% порівняно зі здоровими добровольцями. У пацієнтів з тяжкими порушеннями функції печінки (>9 балів за класифікацією Чайлда — П’ю) експозиція була значно нижча (на 44%).
Порушення функції нирок. Експозиція ліраглутиду була знижена у пацієнтів з порушеннями функції нирок порівняно з особами з нормальною функцією нирок у процесі дослідження із застосуванням одноразової дози (0,75 мг). У пацієнтів з легкими порушеннями (кліренс креатиніну 50–80 мл/хв) експозиція знижувалася на 33%, з порушеннями помірної тяжкості (кліренс креатиніну 30–50 мл/хв) — на 14%, з тяжкими порушеннями (кліренс креатиніну <30 мл/хв) — на 27%, а на кінцевих стадіях захворювань нирок, що вимагають проведення діалізу, — на 26%.
Діти. У клінічних дослідженнях фармакокінетичних властивостей брали участь пацієнти з ожирінням віком 12–17 років (14 пацієнтів з масою тіла 80-122 кг) та 7-11 років (16 пацієнтів з масою тіла 45-87 кг). Експозиція ліраглутиду у підлітків (віком 12–17 років) була подібною до експозиції у дорослих з надмірною масою тіла. Порівнювалась експозиція при введенні 3,0 мг ліраглутиду у дорослих, підлітків та дітей віком 7-11 років після корекції маси тіла.
Доклінічні дані з безпеки. Доклінічні дані, що базуються на дослідженнях з фармакологічної безпеки, токсичності повторних доз та генотоксичності, не виявили жодного ризику для людини. У процесі дворічних досліджень канцерогенності у щурів та мишей були виявлені пухлини С-клітин щитовидної залози, що не призводили до летального результату. Нетоксична доза (NOAEL) у щурів не була встановлена. У мавп, що отримували лікування протягом 20 місяців, таких пухлин не виявлено. Пухлини у гризунів обумовлені негенотоксичним специфічним ГПП–1–рецептор-опосередкованим механізмом, до якого частково чутливі гризуни. Значущість цього механізму у людей достатньо низька, але не може бути повністю виключена. Розвитку інших пухлин не було виявлено. У процесі експериментів на тваринах не було виявлено прямого шкідливого впливу на фертильність, проте при введенні найвищих доз відзначалося незначне підвищення ранньої ембріональної летальності. Введення ліраглутиду в період середини вагітності спричиняло зниження маси тіла самки, уповільнення росту плода з нез’ясованим впливом на розвиток ребер у щурів і скелета у кроликів. При введенні ліраглутиду відзначено уповільнення росту новонароджених щурів, що зберігається в період відлучення від годування молоком у групі прийому високої дози. Невідомо, чи уповільнення росту новонароджених щурів обумовлене зниженням споживання ними молока в результаті прямого впливу ГПП–1, чи зменшенням молока у матері, що обумовлено зниженням калорійності споживаної їжі.
ПОКАЗАННЯ:
лікарський засіб Саксенда® застосовують для зменшення маси тіла як доповнення до дієти зі зниженою калорійністю та збільшеною фізичною активністю у дорослих пацієнтів з початковим індексом маси тіла (ІМТ) більше 30 кг/м2 (ожиріння) або від 27 до 30 кг/м2 (надмірна маса тіла) за наявності хоча б одного супутнього захворювання, пов’язаного з масою тіла, такого як дисглікемія (переддіабет або цукровий діабет 2 типу), гіпертензія, дисліпідемія або обструктивне апное сну.Якщо через 12 тижнів після прийому добової дози 3,0 мг хворий не втратив щонайменше 5% від початкової маси тіла, застосування лікарського засобу Саксенда® слід припинити.
ЗАСТОСУВАННЯ:
дозування
Початкова доза становить 0,6 мг на добу. Для поліпшення переносимості з боку шлунково-кишкового тракту дозу слід збільшувати щотижня на 0,6 мг до досягнення добової дози 3,0 мг (див. таблицю 1). У разі поганої переносимості підвищеної дози протягом двох тижнів слід розглянути питання про припинення лікування. Добова доза вище 3,0 мг не рекомендується.Таблиця 1Графік збільшення дози
| | Доза, мг | Тижні |
| Збільшення дози протягом 4 тижнів | 0,6 | 1 |
| 1,2 | 1 |
| 1,8 | 1 |
| 2,4 | 1 |
| Підтримуюча доза | 3,0 мг |
|
Пропущена доза
Якщо ін’єкція пропущена протягом 12 год з моменту її звичайного введення, пацієнт повинен прийняти дозу якнайшвидше. Якщо до наступного введення залишається менше 12 год, пацієнт не повинен приймати пропущену дозу, а продовжувати прийом 1 раз на добу — прийняти наступну заплановану дозу препарату. Не слід приймати додаткову дозу чи збільшувати дозу для компенсації пропущеної ін’єкції.
Пацієнти з цукровим діабетом 2 типу
Не слід застосовувати лікарський засіб Саксенда® разом з іншими агоністами рецепторів ГПП-1. Для попередження виникнення гіпоглікемії перед застосуванням лікарського засобу Саксенда® слід розглянути можливість зменшення дози стимуляторів секреції інсуліну (наприклад, сульфонілсечовини) або інсуліну. Необхідний контроль рівня глюкози в крові для коригування дози інсуліну чи стимуляторів секреції інсуліну (див. ОСОБЛИВОСТІ ЗАСТОСУВАННЯ).
Особливі групи пацієнтів
Пацієнти літнього віку (≥65 років). Корекція дози у зв’язку з віком не потрібна. Досвід застосування препарату пацієнтам ≥75 років обмежений, тому не рекомендовано застосовувати його даній категорії пацієнтів (див. Фармакокінетика та ОСОБЛИВОСТІ ЗАСТОСУВАННЯ).
Пацієнти з порушеннями функції нирок
Корекція дози не потрібна у пацієнтів з легким або середнім ступенем порушення функції нирок (кліренс креатиніну ≥30 мл/хв). Не рекомендується застосовувати лікарський засіб Саксенда® пацієнтам з тяжкими порушеннями функції нирок (кліренс креатиніну <30 мл/хв), включаючи пацієнтів із термінальною стадією порушення функції нирок (див. Фармакокінетика, ОСОБЛИВОСТІ ЗАСТОСУВАННЯ та ПОБІЧНА ДІЯ).
Пацієнти з порушеннями функції печінки
Не рекомендується коригування дози пацієнтам з легким або середнім ступенем порушення функції печінки. Застосування лікарського засобу Саксенда® не рекомендується пацієнтам з вираженою печінковою недостатністю, а пацієнтам із легкими або помірними порушеннями печінки слід застосовувати його з обережністю (див. Фармакокінетика та ОСОБЛИВОСТІ ЗАСТОСУВАННЯ).
Спосіб введення
Лікарський засіб Саксенда® призначений тільки для підшкірного введення. Його не можна вводити в/в або в/м. Препарат вводять підшкірно 1 раз на добу у будь-який час незалежно від вживання їжі. Його можна вводити підшкірно в ділянку передньої черевної стінки, стегна або плеча. Місце і час введення можна змінювати без корекції дози, проте бажано вводити приблизно в один і той же найбільш зручний час. Для подальшої інформації щодо введення див. розділ Інструкція із використання шприц–ручки.
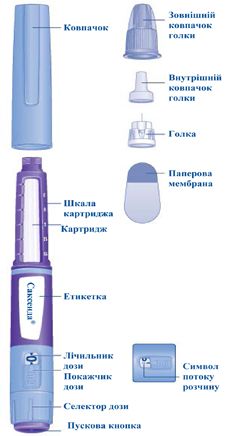
Інструкція з використання шприц–ручки Саксенда®, 6 мг/мл, розчин для ін’єкцій у попередньо заповненій шприц–ручці
Необхідно уважно причитати інструкцію перед використанням шприц–ручки Саксенда®. Не використовувати шприц–ручку без отримання належної інформації щодо її використання від лікаря чи медсестри.Застосування препарату необхідно почати з перевірки шприц–ручки, щоб бути впевненим, що вона містить саме лікарський засіб Саксенда®, 6 мг/мл, потім потрібно подивитися рисунки нижче, щоб дізнатися про різні частини шприц–ручки та голки.Якщо в пацієнта поганий зір або він не бачить взагалі, можна застосовувати шприц–ручку без сторонньої допомоги. Допомагати має людина з хорошим зором, яка може побачити лічильник дози на шприц–ручці Саксенда® та яка вміє користуватися нею.Шприц–ручка Саксенда® є попередньо заповненою. Вона містить 18 мг ліраглутиду, що дає змогу ввести дози 0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг та 3,0 мг. Шприц–ручка Саксенда® призначена для використання з одноразовими голками НовоФайн® або НовоТвіст довжиною до 8 мм і товщиною 32G. Голки не входять у комплект.
Важлива інформація
Необхідно звернути особливу увагу на цю позначку, оскільки вона є важливою для безпечного користування шприц-ручкою.Шприц-ручка Саксенда® та голка (приклад)
| 1. Підготовка шприц-ручки з новою голкою для використання
• Перевірте назву та кольорову етикетку Вашої шприц-ручки, щоб бути впевненим, що вона містить лікарський засіб Саксенда®. Це особливо важливо в тому випадку, якщо Ви застосовуєте різні ін’єкційні лікарські засоби. Застосування неправильного лікарського засобу може бути шкідливим для Вашого здоров’я. • Зніміть ковпачок шприц-ручки.
|  |
|
| Переконайтеся, що розчин у шприц-ручці прозорий та безбарвний. Подивіться у вікно шкали картриджа. Якщо препарат мутний, шприц-ручку використовувати заборонено. | 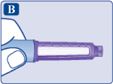 |
| • Візьміть нову одноразову голку та видаліть з неї захисну мембрану. |  |
| • Нагвинтіть голку на шприц-ручку та поверніть її, щоб голка щільно трималась на шприц-ручці. |  |
| • Зніміть зовнішній ковпачок голки та збережіть його. Він знадобиться після завершення ін’єкції для безпечного зняття голки. | 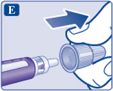 |
| Зніміть внутрішній ковпачок голки та викиньте його. Якщо Ви спробуєте надіти внутрішній ковпачок знову на голку, то можете поранитись. На кінці голки може з’явитися крапля розчину. Це нормальне явище, проте необхідно перевірити надходження препарату при використанні нової шприц-ручки вперше.Не приєднуйте нову голку до тих пір, поки не будете готові зробити ін’єкцію.∎ Для кожної ін’єкції завжди використовуйте нову голку. Це зменшить ризик закупорки голки, зараження, потрапляння інфекції та введення неправильної дози препарату.∎ Ніколи не використовуйте голку, якщо вона погнута чи пошкоджена. | 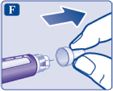 |
| 2. Перевірка роботи шприц-ручки
• Перед першим введенням кожної нової шприц-ручки перевіряйте потік. Якщо Ви вже використовували цю шприц-ручку, переходьте до пункту 3.• Повертайте селектор дози, поки лічильник дози не покаже символ перевірки потоку. |  |
| • Тримайте шприц-ручку голкою вгору. Натисніть і потримайте кнопку дози, поки лічильник дози не повернеться до 0. Значення 0 повинно відповідати показнику дози. На кінчику голки повинна з’явитися крапля розчину.
На кінчику голки може залишитися невелика крапля, але вона не буде вводитися. Якщо жодна крапля не з’явилася, повторіть крок 2 «Перевірка роботи шприц-ручки» до 6 разів. Якщо краплі все ще немає, змініть голку і повторіть крок 2 «Перевірка роботи шприц-ручки» ще раз. Якщо крапля все-таки не з’явилася, утилізуйте ручку і використовуйте нову. ∎ Потрібно завжди переконуватись, що крапля з’являється на кінчику голки, перш ніж вперше використовувати нову ручку. Це гарантує, що розчин буде введений.Якщо крапля не з’явиться, не використовуйте шприц-ручку, навіть якщо лічильник дози змінює значення. Це може вказувати на заблоковану або пошкоджену голку.Якщо Ви не перевірите потік перед першою ін’єкцією нової ручки, то можете не отримати необхідну дозу розчину для забезпечення бажаної дії лікарського засобу.
|  |
| 3. Виставлення дози
Повертайте селектор дози, поки лічильник дози не покаже необхідну дозу (0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг або 3,0 мг). Якщо Ви вибрали неправильну дозу, можна повернути селектор дози вперед або назад до правильної дози.Шприц-ручка вміщує максимум 3,0 мг препарату.За допомогою селектора можна змінити дозу. Тільки лічильник дози та показник дози покажуть, яку кількість міліграмів Ви вибираєте для ін’єкції. Якщо шприц-ручка містить менше 3,0 мг, лічильник дози зупиняється до значення 3,0 мг. При повертанні селектора вперед або назад чутно різне клацання. Не рахуйте клацання.∎ Завжди використовуйте лічильник дози та показник дози, щоб побачити, скільки міліграмів Ви вибрали перед введенням цього препарату.Не використовуйте шкалу картриджа. Вона показує приблизну кількість розчину, яка залишилась у шприц-ручці.Повинні бути виставлені лише дози 0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг або 3,0 мг. Цифри на дисплеї повинні точно збігатися з показником дози, щоб забезпечити правильну дозу для введення. |  |
| Залишок розчину в шприц-ручці• Шкала шприц-ручки показує приблизно, скільки розчину залишилось у шприц-ручці. |  |
| • Щоб точно побачити, скільки розчину залишилось, скористайтеся лічильником дози:Повертайте селектор дози вліво, поки лічильник дози не зупиниться.Якщо він показує 3,0 мг, у шприц-ручці залишилося щонайменше 3,0 мг. Якщо лічильник дози зупиняється до 3,0 мг, значить у шприц-ручці не вистачає розчину для повної дози 3,0 мг.Якщо Вам потрібна вища доза, ніж та, що залишилася у Вашій шприц-ручці. Ви можете розділити свою дозу між Вашою поточною шприц-ручкою та новою шприц-ручкою тільки за умови, що Вас проінструктував лікар або медсестра. Будьте дуже обережні, щоб правильно розрахувати дозу.∎ Будьте дуже уважні, розраховуючи дозу.Якщо Ви не впевнені, як розділити дозу за допомогою двох шприц-ручок, виберіть і введіть необхідну дозу новою шприц-ручкою.
|  |
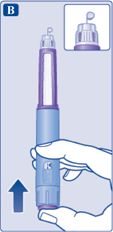
| 4. Введення дози
• Введіть голку під шкіру так, як показав Вам лікар чи медсестра.• Переконайтеся, що Ви бачите лічильник дози. Не закривайте лічильник пальцями. |  |
| • Натисніть і утримуйте пускову кнопку, поки лічильник дози не покаже 0. 0 повинен вирівнюватися вказівником дози. Потім Ви можете почути клацання. |  |
| • Тримайте голку в шкірі після того, як лічильник дози повернеться до 0 і рахуйте повільно до 6. • Якщо голку витягти раніше, Ви можете побачити потік розчину з кінчика голки. В такому випадку правильна доза не буде введена. |  |
| • Витягніть голку з-під шкіри. Якщо крапля крові з’явилась у місці ін’єкції, легенько притисніть це місце, але не розтирайте його. Ви можете побачити краплю розчину на кінчику голки після введення. Це нормально і не впливає на об’єм введеної дози.∎ Завжди дивіться на покажчик дози, щоб бачити скільки міліграмів розчину Ви ввели. Тримайте натиснутою пускову кнопку, поки покажчик дози не покаже 0.
Як виявити заблоковану чи пошкоджену голку?
•Якщо 0 не відображається на лічильнику дози після постійного натискання пускової кнопки, можливо Ви використали заблоковану або пошкоджену голку. •У цьому випадку Ви не ввели потрібну кількість лікарського засобу, навіть незважаючи на те, що лічильник дози перемістився від раніше встановленої дози. Як поводитися із заблокованою голкою?Зніміть голку як описано в пункті 5. «Після ін’єкції» і повторіть усі кроки, починаючи з Кроку 1 «Підготовка шприц-ручки з новою голкою для використання»Ніколи не торкайтеся лічильника дози при введенні. Це може перервати ін’єкцію. |  |
| 5. Після ін’єкції
Вдягніть зовнішній ковпачок на голку, не торкаючись голки або зовнішньої кришки голки. |  |
| Як тільки голка накривається, обережно повністю натисніть на зовнішню кришку голки. Обережно викрутіть голку та утилізуйте. |  |
| • Закривайте кришкою шприц-ручку після кожного використання, щоб захистити розчин від світла.
• Завжди утилізуйте голку після кожної ін'єкції, щоб забезпечити зручність ін’єкції та запобігти заблокуванню голки. Якщо голка заблокована, Ви не зможете ввести препарат. Коли ручка порожня, викиньте її без голки за інструкціями, отриманими від лікаря, медсестри, фармацевта відповідно до місцевих правил.∎ Ніколи не намагайтеся надіти внутрішню кришку голки назад на голку. Ви можете поранитися голкою. ∎ Завжди виймайте голку з ручки після кожної ін’єкції. Це може запобігти заблокуванню голки, забрудненню, зараженню, протіканню розчину та неточному дозуванню.
|  |
| Важлива інформація
Завжди тримайте шприц-ручку та голки в недоступному місці для інших людей, особливо дітей. Ніколи не передавайте Вашу особисту шприц-ручку чи голки іншим людьми. Люди, що доглядають, мають бути дуже обережними при роботі з використаними голками, щоб запобігти травмуванню голкою та перехресному зараженню.
| |
| Догляд за Вашою шприц-ручкою
Не залишайте ручку в машині чи іншому місці, де вона може нагрітися або занадто охолодитись. Не вводити заморожений препарат. У разі введення раніше замороженого препарату Саксенда® Ви можете не отримати очікуваного лікувального ефекту від цього препарату. Зберігайте шприц-ручку від пилу, бруду та рідин. Не мийте, не замочуйте та не змащуйте шприц-ручку. При необхідності очистіть її за допомогою тканини, змоченої м’яким миючим засобом. Запобігайте падінню та ударам шприц-ручки об тверді поверхні. В разі падіння чи удару приєднайте нову голку і перевірте подачу розчину перед введенням. Не намагайтеся наповнити шприц-ручку після закінчення в ній лікарського засобу. Утилізуйте шприц-ручку після закінчення в ній розчину. Не намагайтеся ремонтувати шприц-ручку чи розкладати її.
| |
|
Діти. Безпека та ефективність застосування лікарського засобу Саксенда® у дітей та підлітків (віком до 18 років) дотепер не визначені. Відомі дані зазначені в розділах Фармакодинаміка, Фармакокінетика та ПОБІЧНА ДІЯ, але рекомендацій щодо застосування лікарського засобу немає.Європейське агентство з лікарських засобів відклало зобов’язання подавати результати досліджень застосування лікарського засобу Саксенда® в одній або декількох підгрупах дитячого населення при лікуванні ожиріння та при лікуванні синдрому Прадера-Віллі (див. ЗАСТОСУВАННЯ).
ПРОТИПОКАЗАННЯ:
підвищена чутливість до діючої речовини або до інших компонентів лікарського засобу.
ПОБІЧНА ДІЯ:
резюме профілю з безпеки
Для оцінки ефектів впливу лікарського засобу Саксенда® на контроль рівня глікемії було проведено п’ять подвійних сліпих рандомізованих плацебо-контрольованих клінічних досліджень, у яких брали участь 5813 пацієнтів з ожирінням, надмірною масою тіла та ті, які мали хоча б одне супутнє захворювання, пов’язане з надмірною масою тіла. Найчастішими побічними реакціями були розлади травної системи (67,9%) (див. Опис окремих побічних реакцій).
Список побічних реакцій
Нижче зазначені побічні реакції, про які повідомлялося. Побічні реакції класифіковані за системами органів та частотою виникнення. Оцінку частоти виникнення побічних реакцій проводили за такою шкалою: дуже часто (≥1/10), часто (від ≥1/100 до <1/10), нечасто (від ≥1/1000 до <1/100), рідко (від ≥1/10000 до <1/1000), дуже рідко (<1/10000). У кожній групі побічні реакції наведені в порядку зниження їх серйозності.
З боку імунної системи: рідко – анафілактичні реакції.
Порушення метаболізму і харчування: часто — гіпоглікемія*; нечасто — зневоднення.
Психічні розлади: часто — безсоння**.
З боку нервової системи: часто — запаморочення, дисгевзія.
З боку серцево-судинної системи: нечасто — тахікардія.З боку травної системи: дуже часто — нудота, блювання, діарея, запор; часто — сухість у роті, диспепсія, гастрит, гастроезофагеальна рефлюксна хвороба, біль у верхньому відділі черевної порожнини, метеоризм, еруктація, здуття живота; нечасто — панкреатит***, затримка випорожнення шлунка****.
З боку печінки та жовчних шляхів: часто — жовчнокам’яна хвороба***; нечасто — холецистит***.
З боку шкіри та підшкірних тканин: нечасто — кропив’янка.
З боку нирок та сечовивідних шляхів: рідко — гостра ниркова недостатність, порушення функції нирок.
Загальні розлади та реакції в місці ін’єкції: часто — реакції в місцях ін’єкцій, астенія, втома; нечасто – нездужання.
Лабораторні дослідження: часто — підвищений рівень ліпази, підвищений рівень амілази.*Гіпоглікемія (базується на симптомах, про які повідомляли самі пацієнти і які не підтверджені вимірюваннями рівня глюкози в крові) виникала у пацієнтів, які не страждають на цукровий діабет 2 типу та які застосовували лікарський засіб Саксенда® у поєднанні з дієтою та фізичною активністю. Для отримання додаткової інформації див. Опис окремих побічних реакцій.**Безсоння, в основному спостерігалося протягом перших 3 місяців лікування.***Див. ВЗАЄМОДІЯ З ІНШИМИ ЛІКАРСЬКИМИ ЗАСОБАМИ.****З контрольованої фази 2, 3а та 3б клінічних досліджень.
Опис окремих побічних реакцій
Гіпоглікемія в пацієнтів, які не страждають на цукровий діабет 2 типу
У процесі клінічних випробувань не було зафіксованого жодного тяжкого випадку виникнення гіпоглікемії (що потребує сторонньої допомоги) у пацієнтів з надмірною масою тіла чи ожирінням, які не страждали на цукровий діабет 2 типу та застосовували лікарський засіб Саксенда® у поєднанні з дієтою та фізичною активністю. Про виникнення гіпоглікемії повідомили 1,6% пацієнтів, які отримували лікарський засіб Саксенда®, та 1,1% пацієнтів, які отримували плацебо. Однак ці випадки не були підтверджені вимірюванням рівня глюкози в крові. Більшість випадків легко переносились.
Гіпоглікемія у хворих на цукровий діабет 2 типу
У процесі клінічних досліджень були повідомлення про випадки тяжкої гіпоглікемії у 0,7% пацієнтів з надмірною масою тіла чи ожирінням, які страждали на цукровий діабет 2 типу й застосовували лікарський засіб Саксенда® у поєднанні з дієтою та фізичною активністю та які застосовували сульфонілсечовину. Також зафіксовано виникнення симптомів гіпоглікемії у 43,6% пацієнтів, які застосовували лікарський засіб Саксенда®, та у 27,3% пацієнтів, які отримували плацебо. Серед пацієнтів, які не застосовували сульфонілсечовину, було зафіксовано виникнення симптомів гіпоглікемії (визначаються концентрацією глюкози у плазмі крові ≤3,9 ммоль/л) у 15,7% пацієнтів, які отримували лікарський засіб Саксенда®, та у 7,6% пацієнтів, які отримували плацебо.
Гіпоглікемія у хворих на цукровий діабет 2 типу, які отримують інсулін
У процесі клінічних досліджень були повідомлення про випадки тяжкої гіпоглікемії у 1,5 % пацієнтів з надмірною масою тіла чи ожирінням, які страждали на цукровий діабет 2 типу та застосовували ліраглутид по 3,0 мг/добу разом з інсуліном у поєднанні з дієтою та фізичною активністю. Серед пацієнтів було зафіксовано виникнення симптомів гіпоглікемії (визначаються концентрацією глюкози в плазмі крові ≤3,9 ммоль/л) у 47,2% пацієнтів, які отримували ліраглутид, та у 51,8% пацієнтів, які отримували плацебо. Серед пацієнтів, які застосовували сульфонілсечовину, були зафіксовані повідомлення про виникнення симптомів гіпоглікемії у 60,9% пацієнтів, які отримували ліраглутид по 3,0 мг/добу, та у 60,0% пацієнтів, які отримували плацебо.
Розлади травної системи
Більшість випадків виникнення розладів травної системи були легкого або помірного ступеня тяжкості та не призводили до припинення терапії. Зазвичай реакції виникали протягом перших тижнів лікування і зменшувались протягом декількох днів або тижнів при продовженні лікування. У пацієнтів віком від 65 років при застосуванні лікарського засобу Саксенда® частіше спостерігатися порушення з боку травної системи.У пацієнтів з порушеннями функції нирок легкої або середньої тяжкості (кліренс креатиніну ≥30 мл/хв) при застосуванні лікарського засобу Саксенда® можуть частіше виникати порушення з боку травної системи.
Гостра ниркова недостатність
Зафіксовано випадки виникнення гострої ниркової недостатності у пацієнтів, які застосовували агоністи рецепторів ГПП-1. Більшість зареєстрованих випадків спостерігали у пацієнтів, які страждали на нудоту, блювання та діарею, що і призводило до втрати рідини (див. ОСОБЛИВОСТІ ЗАСТОСУВАННЯ).
Алергічні реакції
Були повідомлення про кілька випадків виникнення анафілактичних реакцій, що супроводжувались такими симптомами як гіпотензія, підвищене серцебиття, напади задухи та набряки після застосування ліраглутиду. Анафілактичні реакції можуть бути небезпечними для життя. Тому якщо є підозра на виникнення анафілактичної реакції, слід припинити застосування ліраглутиду (див. ПРОТИПОКАЗАННЯ).
Реакції в місці ін’єкції
Повідомлялось про реакції в місці введення лікарського засобу Саксенда®. Ці реакції зазвичай були легкими, більшість із них зникали в процесі подальшого лікування.
Тахікардія
У процесі клінічних досліджень були повідомлення про випадки тахікардії у 0,6% пацієнтів, які отримували лікарський засіб Саксенда®, та у 0,1% пацієнтів, які отримували плацебо. Більшість випадків були легкого або помірного ступеня тяжкості. Випадки були поодинокими, більшість із них зникали в процесі подальшого лікування.
Діти
Лікарський засіб Саксенда® не рекомендується для застосування дітям. У двох завершених дослідженнях найчастіше спостерігалися побічні реакції з боку травної системи.
ОСОБЛИВОСТІ ЗАСТОСУВАННЯ:
Спостереження
З метою покращення спостереження за біологічними лікарськими засобами, назва та номер серії препарату, що вводиться. повинен бути чітко зазначений.
Серцева недостатність
Немає терапевтичного досвіду лікування пацієнтів із застійною серцевою недостатністю IV класу за класифікацією Нью-Йоркської асоціації кардіологів (NYHA), тому ліраглутид не рекомендовано застосовувати цим пацієнтам.
Особливі групи пацієнтів
Безпека та ефективність застосування ліраглутиду не встановлені у пацієнтів:— віком ≥75 років;— які застосовують інші лікарські засоби для корекції маси тіла;— із вторинним ожирінням, викликаним ендокринологічними розладами чи розладами, пов’язаними з харчуванням, або в результаті застосування лікарських засобів, що можуть спричинити збільшення маси тіла;— з тяжкими порушеннями функції нирок;— з тяжкими порушеннями функції печінки.Не рекомендується застосовувати лікарський засіб Саксенда® даним групам пацієнтів (див. Дозування). Оскільки дослідження щодо застосування ліраглутиду для корекції маси тіла в пацієнтів з легкими або помірними порушеннями функції печінки відсутні, його слід з обережністю застосовувати цій групі пацієнтів (див. Фармакокінетика та Дозування).Досвід застосування ліраглутиду хворим із запальними захворюваннями кишечнику і діабетичним гастропарезом обмежений. Застосування ліраглутиду цим пацієнтам не рекомендовано, оскільки воно супроводжується тимчасовими побічними реакціями з боку шлунково-кишкового тракту, в т. ч. нудотою, блюванням і діареєю.
Панкреатит
Спостерігались випадки гострого панкреатиту при застосуванні аналогів рецептора ГПП-1. Пацієнтів слід проінформувати про характерні симптоми гострого панкреатиту. При підозрі на панкреатит слід відмінити лікування ліраглутидом. Якщо підтверджується гострий панкреатит, повторне застосування ліраглутиду не рекомендоване.
Жовчнокам’яна хвороба та холецистит
У клінічних випробуваннях при застосуванні ліраглутиду для зменшення маси тіла відсоток виникнення жовчнокам’яної хвороби та холециститу був вищий порівняно з пацієнтами, які отримували плацебо. Той факт, що швидка втрата маси тіла може збільшити ризик розвитку жовчнокам’яної хвороби і, отже, холециститу, лише частково пояснює більш високу частоту виникнення даних захворювань після застосування ліраглутиду. Жовчнокам’яна хвороба та холецистит можуть призвести до госпіталізації та холецистектомії. Пацієнтів слід поінформувати про характерні симптоми жовчнокам’яної хвороби та холециститу.
Захворювання щитовидної залози
У процесі клінічних досліджень цукрового діабету 2 типу відмічені побічні реакції з боку щитовидної залози, такі як зоб, особливо у пацієнтів з уже наявними захворюваннями щитовидної залози. Тому ліраглутид слід з обережністю застосовувати цим пацієнтам.
Частота серцевих скорочень
Під час клінічних досліджень ліраглутиду спостерігалося збільшення частоти серцевих скорочень (див. Фармакодинаміка). Частоту серцевих скорочень слід контролювати через рівні проміжки часу. Пацієнти мають бути проінформовані про симптоми збільшення частоти серцебиття (підвищене серцебиття або відчуття підвищеного серцебиття в спокої). Пацієнтам, у яких спостерігається клінічно стійке значне збільшення частоти серцевих скорочень у спокої, лікування ліраглутидом слід припинити.
Зневоднення
У хворих, які застосовували агоністи рецепторів ГПП-1, спостерігалися симптоми зневоднення, в тому числі порушення функції нирок та гострої ниркової недостатності. Пацієнтів, яким призначено ліраглутид, необхідно проінформувати про можливість зневоднення організму внаслідок розладів травної системи та необхідність вживання запобіжних заходів щодо зневоднення.
Гіпоглікемія у хворих на цукровий діабет 2 типу
У пацієнтів із цукровим діабетом 2 типу, які отримують ліраглутид у поєднанні з інсуліном та/або сульфонілсечовиною, може бути підвищений ризик виникнення гіпоглікемії. Ризик виникнення гіпоглікемії може бути знижений з допомогою зменшення дози інсуліну та/або сульфонілсечовини.
Гіперглікемія у хворих на цукровий діабет 2 типу, які отримують інсулін
Лікарський засіб Саксенда® не застосовують як замінник інсуліну хворим на цукровий діабет. Повідомлялося про розвиток діабетичного кетоацидозу у пацієнтів, які застосовували інсулін, у разі швидкого припинення або зменшення дози як замінника інсуліну (див. ЗАСТОСУВАННЯ).
Допоміжні речовини
Саксенда® містить менше ніж 1 ммоль натрію (23 мг), тому лікарський засіб можна вважати таким, що не містить натрію.
Застосування у період вагітності або годування груддю.
Вагітність. Адекватні дані щодо застосування ліраглутиду вагітним жінкам відсутні. Дослідження на тваринах показали репродуктивну токсичність (див. Доклінічні дані з безпеки). Потенційний ризик для людей невідомий. Ліраглутид не слід застосовувати під час вагітності. Якщо пацієнтка хоче завагітніти або вагітна, то прийом ліраглутиду необхідно відмінити.
Період годування груддю. Невідомо, чи екскретується ліраглутид у грудне молоко людини. Дослідження на тваринах показали, що в молоко потрапляє незначна кількість ліраглутиду і його близькоспоріднених структурних метаболітів. Доклінічні дослідження виявили пов’язане із застосуванням препарату зменшення темпів зростання новонароджених щуренят (див. Доклінічні дані про безпеку застосування). У зв’язку з недостатнім досвідом застосування препарату у період годування груддю не слід застосовувати його в цей період.
Фертильність. Окрім незначного зменшення кількості живих імплантованих ембріонів, дослідження на тваринах не виявили шкідливого впливу препарату на репродуктивну здатність (див. Доклінічні дані про безпеку застосування).
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами. Лікарський засіб Саксенда® не впливає або має незначний вплив на здатність керувати транспортними засобами й іншими механізмами. Однак може виникати запаморочення здебільшого протягом перших 3 місяців лікування лікарським засобом Саксенда®. Слід з обережністю керувати автотранспортом або іншими механізмами при виникненні запаморочення.
ВЗАЄМОДІЯ З ІНШИМИ ЛІКАРСЬКИМИ ЗАСОБАМИ:
Іn vitro ліраглутид продемонстрував дуже низький потенціал впливу на фармакокінетику інших активних субстанцій, обмін яких пов’язаний із цитохромом Р450, а також зв’язування з білками плазми крові. Ліраглутид спричинює незначну затримку випорожнення шлунка, що може вплинути на всмоктування пероральних препаратів, що застосовуються одночасно. Дослідження щодо взаємодії не показали будь-якого клінічно значущого уповільнення всмоктування, тому корекція дози не потрібна. Дослідження взаємодії проводили при застосуванні ліраглутиду в дозі 1,8 мг. Вплив на швидкість випорожнення шлунка був еквівалентним до ліраглутиду в дозі 1,8 мг та 3,0 мг (парацетамол AUC0-300 хв). Зареєстрований щонайменше один епізод виникнення гострої діареї у деяких пацієнтів, які отримували лікарський засіб Саксенда®. Діарея може порушувати всмоктування пероральних лікарських засобів, що одночасно приймаються.
Варфарин та інші похідні кумарину. Досліджень лікарської взаємодії не проводили. Не можна виключити клінічно значущу взаємодію з активною субстанцією, що має низьку розчинність або вузький терапевтичний індекс, такою як варфарин. На початку лікування ліраглутидом у пацієнтів, які одержують варфарин або інші похідні кумарину, рекомендується частіше проводити контроль міжнародного нормалізованого співвідношення (МНС).
Парацетамол. Ліраглутид не змінював загальну експозицію парацетамолу після введення одноразової дози 1000 мг. Максимальна концентрація парацетамолу (Cmax) знижувалася на 31%, а час досягнення максимальної концентрації (tmax) збільшувався до 15 хв. При одночасному застосуванні парацетамолу корекція дози не потрібна.
Аторвастатин. Ліраглутид не змінював загальну експозицію аторвастатину до клінічно значущого рівня після одноразового його введення в дозі 40 мг. У зв’язку з цим при одночасному застосуванні з ліраглутидом корекція дози аторвастатину не потрібна. При одночасному введенні з ліраглутидом Cmax аторвастатину знижувалася на 38%, а tmax збільшувався з 1 год до 3 год.
Гризеофульвін. Ліраглутид не змінював загальної експозиції гризеофульвіну після одноразового його введення в дозі 500 мг. Cmax гризеофульвіну зростала на 37%, тоді як tmax не змінювався. Коригування дози при застосуванні гризеофульвіну та інших низькорозчинних сполук з високою проникністю не потрібне.
Дигоксин. Після одноразового введення 1 мг дигоксину у поєднанні з ліраглутидом відмічено зменшення значення AUC для дигоксину на 16%; Cmax знижувалася на 31%. Середній tmax дигоксину збільшувався з 1 год до 1,5 год. Виходячи з даних результатів, корекція дози дигоксину не потрібна.
Лізиноприл. Після одноразового введення 20 мг лізиноприлу відмічено зменшення AUC для лізиноприлу на 15%, Cmax знижувалася на 27%. Середній tmax лізиноприлу збільшувався з 6 до 8 год. Виходячи з даних результатів, корекція дози лізиноприлу не потрібна.
Пероральні контрацептиви. При одночасному застосуванні разової дози пероральних контрацептивів ліраглутид знижував Cmax етинілестрадіолу або левоноргестрелу на 12% і 13% відповідно, а tmax збільшувався на 1,5 год. Це не мало клінічно значущого впливу на загальну експозицію етинілестрадіолу або левоноргестрелу, що дає підставу вважати, що одночасний прийом їх з ліраглутидом не вплине на контрацептивний ефект етинілестрадіолу та левоноргестрелу.
Несумісність. Додавання до лікарського засобу Саксенда® будь-якої субстанції може призвести до деградації ліраглутиду. Оскільки дослідження сумісності не проводилися, цей лікарський засіб не можна застосовувати з іншими лікарськими засобами.
ПЕРЕДОЗУВАННЯ:
у клінічних дослідженнях та повідомленнях, що надійшли після виведення лікарського засобу Саксенда® на ринок, відзначено випадки перевищення рекомендованої підтримувальної дози до 24 разів (72 мг). Загалом пацієнти повідомляли про сильну нудоту, блювання та діарею, але ніхто з пацієнтів не повідомляв про тяжку гіпоглікемію. Усі хворі одужали без ускладнень.При передозуванні слід проводити підтримуюче лікування відповідно до наявних у пацієнта клінічних ознак і симптомів. Необхідне спостереження за клінічними ознаками на випадок виникнення зневоднення та гіпоглікемії.
УМОВИ ЗБЕРІГАННЯ:
зберігати в недоступному для дітей місці. Зберігати в холодильнику (2–8 °С) подалі від морозильної камери. Після першого застосування зберігати при температурі нижче 30 °С або в холодильнику (2–8 °С). Не заморожувати.Для запобігання дії світла зберігати шприц-ручку із закритим ковпачком.
Термін придатності. 30 місяців.Після першого застосування – 1 місяць.
Дата останнього перегляду: 23.03.2021
ІНСТРУКЦІЯ
для медичного застосування лікарського засобу
САКСЕНДА®
(SAXENDA®)
Склад
діюча речовина: ліраглутид;
1 мл розчину містить 6 мг ліраглутиду — аналога людського глюкагоноподібного пептиду-1 (ГПП-1), виготовленого за технологією рекомбінантної ДНК в Saccharomyces cerevisiae;
одна попередньо заповнена шприц-ручка містить 18 мг ліраглутиду в 3 мл;
допоміжні речовини: натрію гідрофосфат, дигідрат; пропіленгліколь; фенол; кислота хлористоводнева (для корекції рН); натрію гідроксид (для корекції рН); вода для ін’єкцій.
Лікарська форма
Розчин для ін’єкцій.
Основні фізико-хімічні властивості: прозорий та безбарвний або майже безбарвний ізотонічний розчин; рН 8,15.
Фармакотерапевтична група
Препарати, що застосовуються при цукровому діабеті, аналоги глюкагоноподібного пептиду-1 (ГПП-1). Код ATХ A10B J02.
Фармакологічні властивості
Фармакодинаміка
Механізм дії
Ліраглутид є ацильованим аналогом ГПП-1 з послідовністю амінокислот, на 97% гомологічною ендогенному людському ГПП-1. Ліраглутид зв’язується з ГПП-1-рецепторами та активує їх.
ГПП-1 є фізіологічним регулятором апетиту та споживання їжі, але точний механізм його дії повністю не встановлений. У дослідженнях на тваринах периферійне введення ліраглутиду призвело до його накопичення в специфічних ділянках мозку, що відповідають за регуляцію апетиту, де ліраглутид завдяки специфічній активації рецепторів ГПП-1 підвищував відчуття насичення і знижував сигнали голоду, що сприяло зниженню маси тіла.
Рецептори ГПП-1 також експресуються у певних ділянках серця, судин, імунній системі та нирках. При моделюванні атеросклерозу у мишей ліраглутид запобігав прогресуванню аортальної атеросклеротичної бляшки та знижував запалення в бляшці. Крім того, ліраглутид позитивно впливав на ліпіди плазми. Ліраглутид не зменшував розмір вже наявних атеросклеротичних бляшок.
Фармакодинамічні ефекти
Ліраглутид знижує масу тіла у людей головним чином за рахунок втрати жирової маси, при цьому завдяки зменшенню переважно вісцерального жиру порівняно з підшкірним. Ліраглутид регулює апетит, підсилюючи відчуття ситості та наповненості шлунка, знижуючи при цьому відчуття голоду, та призводить до зниження споживання їжі. Ліраглутид не збільшує енерговитрати порівняно з плацебо.
Ліраглутид стимулює секрецію інсуліну та зменшує надмірно високу секрецію глюкагону залежно від рівня глюкози, що призводить до зниження глюкози натще та після вживання їжі.
У пацієнтів з переддіабетом та цукровим діабетом ефект зниження рівня глюкози більш виражений порівняно з пацієнтами з нормоглікемією. Клінічні випробування свідчать, що ліраглутид покращує та підтримує функцію бета-клітин відповідно до НОМА-В та співвідношення проінсулін/інсулін.
Клінічна ефективність та безпека
Клінічна ефективність та безпека застосування ліраглутиду для зниження маси тіла як доповнення до дієти зі зниженою калорійністю та збільшеною фізичною активністю були вивчені у чотирьох рандомізованих подвійно сліпих плацебо-контрольованих дослідженнях фази 3 за участю 5358 пацієнтів.
Дослідження 1 (SCALE Ожиріння та переддіабет — 1839)
Всього 3731 пацієнт із ожирінням (індекс маси тіла (ІМТ) ≥ 30 кг/м2) або з надмірною масою тіла (ІМТ ≥ 27 кг/м2) і дисліпідемією та/або артеріальною гіпертензією були стратифіковані за допомогою скринінгу відповідно до статусу переддіабету та початкового рівня ІМТ (≥ 30 кг/м2 або < 30 кг/м2). Всі пацієнти (3731) були рандомізовані на лікування протягом 56 тижнів, а 2254 пацієнти, які мали переддіабет на момент скринінгу, були рандомізовані на лікування протягом 160 тижнів. Обидва періоди лікування супроводжувались 12-тижневим періодом спостереження за групами препарат/плацебо. Корекція способу життя у вигляді дієти зі зниженою калорійністю та збільшеною фізичною активністю була базовою терапією для всіх пацієнтів.
У 56-тижневому дослідженні 1 оцінювали зниження маси тіла у всіх (3731) рандомізованих пацієнтів (2590 пацієнтів завершили курс лікування).
У 160-тижневому дослідженні 1 оцінювали час до розвитку цукрового діабету 2 типу у 2254 рандомізованих пацієнтів з переддіабетом (1128 пацієнтів завершили курс лікування).
Дослідження 2 (SCALE Діабет — 1922)
Дослідження тривалістю 56 тижнів, у якому оцінювали зниження маси тіла у 846 рандомізованих пацієнтів із ожирінням та надмірною масою тіла (628 пацієнтів завершили курс лікування), які мали недостатньо контрольований цукровий діабет 2 типу (HbA1c в діапазоні 7–10%). Основним методом лікування на початку дослідження була або дієта, або збільшена фізична активність, або застосування окремих лікарських засобів, таких як метформін, сульфонілсечовина та глітазон, або їх комбінацій.
Дослідження 3 (SCALE Апное під час сну — 3970)
Дослідження тривалістю 32 тижні, в якому оцінювали ступінь тяжкості апное під час сну та зниження маси тіла у 359 рандомізованих пацієнтів (276 пацієнтів завершили курс лікування) з ожирінням та обструктивним апное під час сну помірного або тяжкого ступеня.
Дослідження 4 (SCALE Підтримуюче лікування — 1923)
У дослідженні тривалістю 56 тижнів оцінювали підтримуюче лікування після зменшення маси тіла на ≥ 5% завдяки дієті зі зниженою калорійністю у 422 рандомізованих пацієнтів із ожирінням та надмірною масою тіла (305 пацієнтів завершили курс лікування) і артеріальною гіпертензією чи дисліпідемією.
Маса тіла
При застосуванні ліраглутиду було досягнуто кращого зниження маси тіла порівняно з плацебо у пацієнтів із ожирінням та надмірною масою тіла у всіх досліджуваних групах. У всіх популяціях дослідження більша частина пацієнтів досягала зниження маси тіла на ≥ 5% та > 10% при застосуванні ліраглутиду порівняно з плацебо (таблиці 1–3). У 160-тижневій частині дослідження 1 зниження маси тіла відбувалося переважно в перший рік терапії і підтримувалося протягом всіх 160 тижнів.
- У дослідженні 4 при застосуванні ліраглутиду більше пацієнтів утримували результат зниження маси тіла, досягнутий до початку лікування, порівняно з плацебо (81,4% та 48,9% відповідно). Конкретні дані досліджень 1–4 щодо зниження маси тіла, терміну лікування та кумулятивного розподілу зміни маси тіла (%) представлені в таблицях 1–5 і на рисунках 1, 2 і 3.
Зниження маси тіла через 12 тижнів при лікуванні ліраглутидом (3,0 мг)
Особами, які мали ранню відповідь на лікування, були пацієнти, які досягли ≥ 5% зниження маси тіла після 12 тижнів застосування терапевтичної дози ліраглутиду (4 тижні ескалації дози та 12 тижнів прийому лікувальної дози). У дослідженні 1 тривалістю 56 тижнів 67,5% пацієнтів досягли ≥ 5% зниження маси тіла після 12 тижнів. У дослідженні 2 50,4% пацієнтів досягли зниження маси тіла ≥ 5% через 12 тижнів. При продовженні лікування прогнозується, що 86,2% пацієнтів досягнуть зниження маси тіла ≥ 5%, а 51% пацієнтів — ≥ 10% після застосування ліраглутиду протягом року. Середнє зниження маси тіла у пацієнтів, які застосовували ліраглутид протягом року, становило 11,2% від їх початкової маси тіла (9,7% для чоловіків і 11,6% для жінок). Серед пацієнтів, які досягли < 5% зниження маси тіла після 12 тижнів терапії, частка пацієнтів, які після року застосування ліраглутиду не досягли зменшення маси тіла ≥ 10%, становила 93,4%.
Контроль глікемії
Лікування ліраглутидом значно покращило показники глікемії у пацієнтів з нормоглікемією, переддіабетом та цукровим діабетом 2 типу. У 56-тижневій частині дослідження 1 цукровий діабет 2 типу розвинувся у меншої кількості пацієнтів, які отримували ліраглутид, порівняно з пацієнтами, які отримували плацебо (0,2% проти 1,1% відповідно). У більшості пацієнтів з переддіабетом на початку лікування спостерігався оборотний розвиток цього захворювання після застосування ліраглутиду порівняно з плацебо (69,2% проти 32,7% відповідно). Первинною кінцевою точкою ефективності у 160-тижневій частині дослідження 1 була частка пацієнтів, у яких виник цукровий діабет 2 типу, оцінена як час до розвитку цукрового діабету. На 160-му тижні дослідження у 3% пацієнтів, які отримували лікарський засіб Саксенда®, і в 11% пацієнтів, які отримували плацебо, діагностували цукровий діабет 2 типу. Розрахунковий час розвитку цукрового діабету 2 типу у пацієнтів, які отримували ліраглутид 3 мг, був у 2,7 рази довший (95% ДІ [1,9; 3,9]), а коефіцієнт ризику розвитку цукрового діабету 2 типу становив 0,2 для ліраглутиду порівняно з плацебо.
Кардіометаболічні фактори ризику
Лікування ліраглутидом значно покращило показники систолічного артеріального тиску та окружності талії порівняно з плацебо (таблиці 1, 2 та 3).
Індекс апное-гіпопное (ІАГ)
При застосуванні ліраглутиду спостерігалось значне зниження тяжкості обструктивного апное під час сну порівняно з плацебо, яке було оцінене за допомогою зменшення порівняно з плацебо (таблиця 4).
Таблиця 1
Дослідження 1: зміни маси тіла, глікемії та кардіометаболічних показників на 56-му тижні порівняно з вихідними величинами
|
Маса тіла
|
Саксенда® (N = 2437)
|
Плацебо (N = 1225)
|
Саксенда® порівняно з плацебо
|
|
Вихідна маса тіла на візиті включення, кг (СВ)
|
106,3 (21,2)
|
106,3 (21,7)
|
-
|
|
Середня зміна на 56-му тижні, % (95 % ДІ)
|
–8,0
|
–2,6
|
–5,4** (–5,8; –5,0)
|
|
Середня зміна на 56-му тижні, кг (95 % ДІ)
|
–8,4
|
–2,8
|
–5,6** (–6,0; –5,1)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на ≥ 5 % на 56-му тижні, % (95 % ДІ)
|
63,5
|
26,6
|
4,8** (4,1; 5,6)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на > 10 % на 56-му тижні, % (95 % ДІ)
|
32,8
|
10,1
|
4,3** (3,5; 5,3)
|
|
Глікемія та кардіометаболічні фактори
|
Вихідний показник на візиті включення
|
Зміна
|
Вихідний показник на візиті включення
|
Зміна
|
|
|
HbA1c, %
|
5,6
|
–0,3
|
5,6
|
–0,1
|
–0,23**
(–0,25; –0,21)
|
|
ГПН, ммоль/л
|
5,3
|
–0,4
|
5,3
|
–0,01
|
–0,38**
(–0,42; –0,35)
|
|
Систолічний артеріальний тиск, мм рт. ст.
|
123,0
|
–4,3
|
123,3
|
–1,5
|
–2,8** (–3,6; –2,1)
|
|
Діастолічний артеріальний тиск, мм рт. ст.
|
78,7
|
–2,7
|
78,9
|
–1,8
|
–0,9* (–1,4; –0,4)
|
|
Окружність талії, см
|
115,0
|
–8,2
|
114,5
|
–4,0
|
–4,2** (–4,7; –3,7)
|
Повна вибірка пацієнтів. Для маси тіла, HbA1c, ГПН, артеріального тиску та окружності талії наведено середні величини, зміни на 56-му тижні порівняно з вихідними показниками є розрахунковими середніми величинами (за методом найменших квадратів), і контрасти комбінацій умов на 56-му тижні є розрахунковими різницями між методами лікування. Для відсотка пацієнтів, маса тіла яких зменшилась на ≥ 5 / > 10%, наведено розрахункове співвідношення шансів. Пропущені значення після вихідного рівня були заміщені шляхом перенесення даних останнього спостереження вперед.
* p < 0,05.
** p < 0,0001.
ДІ — довірчий інтервал.
ГПН — глюкоза в плазмі натще.
СВ — стандартне відхилення.
Таблиця 2
Дослідження 1: зміни маси тіла, глікемії та кардіометаболічних показників на 160-му тижні порівняно з вихідними величинами
|
Маса тіла
|
Саксенда®
(N = 1472)
|
Плацебо (N = 738)
|
Саксенда® порівняно з плацебо
|
|
Вихідна маса тіла на візиті включення, кг (СВ)
|
107,6 (21,6)
|
108,0 (21,8)
|
|
|
Середня зміна на 160-му тижні,% (95% ДІ)
|
-6,2
|
-1,8
|
-4,3**
(–4,9; –3,7)
|
|
Середня зміна на 160-му тижні, кг (95% ДІ)
|
-6,5
|
-2,0
|
-4,6**
(–5,3; –3,9)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на ≥ 5% на 160-му тижні,% (95% ДІ)
|
49,6
|
23,4
|
3,2**
(2,6; 3,9)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на > 10% на 160-му тижні,% (95% ДІ)
|
24,4
|
9,5
|
3,1**
(2,3; 4,1) |
|
Глікемія та кардіометаболічні фактори
|
Вихідний показник на візиті включення
|
Зміна
|
Вихідний показник на візиті включення
|
Зміна
|
|
|
HbA1c,%
|
5,8
|
-0,4
|
5,7
|
-0,1
|
-0,21**
(–0,24; –0,18)
|
|
ГПН, ммоль/л
|
5,5
|
-0,4
|
5,5
|
0,04
|
-0,4**
(–0,5; –0,4)
|
|
Систолічний артеріальний тиск, мм рт. ст.
|
124,8
|
-3,2
|
125,0
|
-0,4
|
-2,8**
(–3,8; –1,8)
|
|
Діастолічний артеріальний тиск, мм рт. ст.
|
79,4
|
-2,4
|
79,8
|
-1,7
|
-0,6 (–1,3; 0,1)
|
|
Окружність талії, см
|
116,6
|
-6,9
|
116,7
|
-3,4
|
-3,5**
(–4,2; –2,8)
|
Повна вибірка пацієнтів. Для маси тіла, HbA1c, ГПН, артеріального тиску та окружності талії наведено середні величини, зміни на 160-му тижні порівняно з вихідними показниками є розрахунковими середніми величинами (за методом найменших квадратів), і контрасти комбінацій умов на 160-му тижні є розрахунковими різницями між методами лікування. Для відсотка пацієнтів, маса тіла яких зменшилась на ≥ 5 / > 10%, наведено розрахункове співвідношення шансів. Пропущені значення після вихідного рівня були заміщені шляхом перенесення даних останнього спостереження вперед.
** p < 0,0001.
ДІ — довірчий інтервал.
ГПН — глюкоза в плазмі натще.
СВ — стандартне відхилення.

Рисунок 1. Зміна маси тіла (%) від початкового показника за часом у дослідженні 1 (0–56 тижнів)

Рисунок 2. Кумулятивний розподіл зміни маси тіла (%) після 56 тижнів лікування у дослідженні 1
Таблиця 3
Дослідження 2: зміни маси тіла, глікемії та кардіометаболічних показників на 56-му тижні порівняно з вихідними величинами
|
Маса тіла
|
Саксенда® (N = 412)
|
Плацебо (N = 211)
|
Саксенда® порівняно з плацебо
|
|
Вихідна маса тіла на візиті включення, кг (СВ)
|
105,6 (21,9)
|
106,7 (21,2)
|
-
|
|
Середня зміна на 56-му тижні, % (95 % ДІ)
|
–5,9
|
–2,0
|
–4,0** (–4,8; –3,1)
|
|
Середня зміна на 56-му тижні, кг (95 % ДІ)
|
–6,2
|
–2,2
|
–4,1** (–5,0; –3,1)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на ≥ 5 % на 56-му тижні, % (95 % ДІ)
|
49,8
|
13,5
|
6,4** (4,1; 10,0)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на > 10 % на 56-му тижні, % (95 % ДІ)
|
22,9
|
4,2
|
6,8** (3,4; 13,8)
|
|
Глікемія та кардіометаболічні фактори
|
Вихідний показник на візиті включення
|
Зміна
|
Вихідний показник на візиті включення
|
Зміна
|
|
|
HbA1c, %
|
7,9
|
–1,3
|
7,9
|
–0,4
|
–0,9** (–1,1; –0,8)
|
|
ГПН, ммоль/л
|
8,8
|
–1,9
|
8,6
|
–0,1
|
–1,8** (–2,1; –1,4)
|
|
Систолічний артеріальний тиск, мм рт. ст.
|
128,9
|
–3,0
|
129,2
|
–0,4
|
–2,6* (–4,6; –0,6)
|
|
Діастолічний артеріальний тиск, мм рт. ст.
|
79,0
|
–1,0
|
79,3
|
–0,6
|
–0,4 (–1,7; 1,0)
|
|
Окружність талії, см
|
118,1
|
–6,0
|
117,3
|
–2,8
|
–3,2** (–4,2; –2,2)
|
Повна вибірка пацієнтів. Для маси тіла, HbA1c, ГПН, артеріального тиску та окружності талії наведено середні величини, зміни на 56-му тижні порівняно з вихідними показниками є розрахунковими середніми величинами (за методом найменших квадратів), і контрасти комбінацій умов на 56-му тижні є розрахунковими різницями між методами лікування. Для відсотка пацієнтів, маса тіла яких зменшилась на ≥ 5 / > 10%, наведено розрахункове співвідношення шансів. Пропущені значення після вихідного рівня були заміщені шляхом перенесення даних останнього спостереження вперед.
* p < 0,05.
** p < 0,0001.
ДІ — довірчий інтервал.
ГПН — глюкоза в плазмі натще.
СВ — стандартне відхилення.
Таблиця 4
Дослідження 3: зміни маси тіла та індексу апное-гіпопное на 32-му тижні порівняно з вихідними величинами
|
Маса тіла
|
Саксенда® (N = 180)
|
Плацебо (N = 179)
|
Саксенда® порівняно з плацебо
|
|
Вихідна маса тіла на візиті включення, кг (СВ)
|
116,5 (23,0)
|
118,7 (25,4)
|
-
|
|
Середня зміна на 32-му тижні, % (95 % ДІ)
|
–5,7
|
–1,6
|
–4,2** (–5,2; –3,1)
|
|
Середня зміна на 32-му тижні, кг (95 % ДІ)
|
–6,8
|
–1,8
|
–4,9** (–6,2; –3,7)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на ≥ 5 % на 32-му тижні, % (95 % ДІ)
|
46,4
|
18,1
|
3,9** (2,4; 6,4)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на > 10 % на 32-му тижні, % (95 % ДІ)
|
22,4
|
1,5
|
19,0** (5,7; 63,1)
|
|
|
Вихідний показник на візиті включення
|
Зміна
|
Вихідний показник на візиті включення
|
Зміна
|
|
|
Індекс апное-гіпопное, епізодів/годину
|
49,0
|
–12,2
|
49,3
|
–6,1
|
–6,1* (–11,0; –1,2)
|
Повна вибірка пацієнтів. Для вихідних показників наведено середні величини, зміни на 32-му тижні порівняно з вихідними показниками є розрахунковими середніми величинами (за методом найменших квадратів), і контрасти комбінацій умов на 32-му тижні є розрахунковими різницями між методами лікування (95% ДІ). Для відсотка пацієнтів, маса тіла яких зменшилась на ≥ 5 / > 10%, наведено розрахункове співвідношення шансів. Пропущені значення після вихідного рівня були заміщені шляхом перенесення даних останнього спостереження вперед.
* p < 0,05.
** p < 0,0001.
ДІ — довірчий інтервал.
СВ — стандартне відхилення.
Таблиця 5
Дослідження 4: зміна маси тіла на 56-му тижні порівняно з вихідною величиною
|
Маса тіла
|
Саксенда® (N = 207)
|
Плацебо (N = 206)
|
Саксенда® порівняно з плацебо
|
|
Вихідна маса тіла на візиті включення, кг (СВ)
|
100,7 (20,8)
|
98,9 (21,2)
|
-
|
|
Середня зміна на тижні 56, % (95 % ДІ)
|
–6,3
|
–0,2
|
–6,1** (–7,5; –4,6)
|
|
Середня зміна на 56-му тижні, кг (95 % ДІ)
|
–6,0
|
–0,2
|
–5,9** (–7,3; –4,4)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на ≥ 5 % на 56-му тижні, % (95 % ДІ)
|
50,7
|
21,3
|
3,8** (2,4; 6,0)
|
|
Відсоток пацієнтів, маса тіла яких зменшилась на > 10 % на 56-му тижні, % (95 % ДІ)
|
27,4
|
6,8
|
5,1** (2,7; 9,7)
|
Повна вибірка пацієнтів. Для вихідних показників наведено середні величини, зміни на 56-му тижні порівняно з вихідними показниками є розрахунковими середніми величинами (за методом найменших квадратів), і контрасти комбінацій умов на 56-му тижні є розрахунковими різницями між методами лікування. Для відсотка пацієнтів, маса тіла яких зменшилась на ≥ 5 / > 10%, наведено розрахункове співвідношення шансів. Пропущені значення після вихідного рівня були заміщені шляхом перенесення даних останнього спостереження вперед.
** p < 0,0001.
ДІ — довірчий інтервал.
СВ — стандартне відхилення.

Рисунок 3. Зміна маси тіла (%) від рандомізації (тиждень 0) за часом у дослідженні 4
До тижня 0 пацієнтів лікували лише за допомогою дієти зі зниженою калорійністю та фізичних вправ. На тижні 0 пацієнти були рандомізовані для застосування або лікарського засобу Саксенда®, або плацебо.
Імуногенність
З огляду на потенційні імуногенні властивості лікарських засобів, що містять білки або пептиди, передбачається, що в пацієнтів можуть утворитися антитіла до ліраглутиду після застосування лікарського засобу Саксенда®. Під час клінічних досліджень у 2,5% пацієнтів, які отримували ліраглутид, утворювались антитіла до ліраглутиду. Утворення антитіл не призводило до зниження ефективності ліраглутиду.
Вплив на серцево-судинну систему
Основні серйозні несприятливі серцево-судинні явища (major adverse cardiovascular events (МАСЕ)), які були визначені зовнішньою незалежною експертною групою: нелетальний інфаркт міокарда, нелетальний інсульт, серцево-судинна смерть. В усіх тривалих клінічних дослідженнях лікарського засобу Саксенда® було зареєстровано 6 МАСЕ у пацієнтів, які отримували ліраглутид, та 10 МАСЕ у пацієнтів, які отримували плацебо. Відношення ризиків та ДІ 95% при порівнянні лікарського засобу Саксенда® та плацебо становило 0,33 [0,12; 0,90].
У клінічних дослідженнях 3-ї фази спостерігалось підвищення частоти серцевих скорочень порівняно з вихідним значенням в середньому на 2,5 удару за хвилину (від 1,6 до 3,6 удару за хвилину в окремих дослідженнях). Максимальне збільшення частоти серцевих скорочень спостерігалося приблизно після 6 тижнів терапії. Тривалий клінічний вплив збільшення частоти серцевих скорочень не встановлений. Це збільшення було оборотним та зникало після припинення терапії ліраглутидом (див. розділ «Особливості застосування»).
У дослідженні LEADER® брали участь 9340 пацієнтів із недостатньо контрольованим цукровим діабетом 2 типу. Переважна кількість із них мали серцево-судинні захворювання. Пацієнтів рандомізовано розподіляли для застосування ліраглутиду у добовій дозі до 1,8 мг (4668) або плацебо (4672) на додаток до стандартного лікування.
Тривалість терапії становила від 3,5 до 5 років. Середній вік пацієнтів становив 64 роки, середній ІМТ — 32,5 кг/м². Середнє значення початкового рівня HbA1c становило 8,7 і покращилося через 3 роки на 1,2% у пацієнтів, яким був призначений ліраглутид, та на 0,8% у пацієнтів, яким було призначено плацебо. Первинною кінцевою точкою ефективності був час від рандомізації до першого виникнення будь-яких основних MACE: нелетального інфаркту міокарда, нелетального інсульту, серцево-судинної смерті. Ліраглутид значно знизив частоту виникнення основних несприятливих серцево-судинних явищ (події первинної кінцевої точки, MACE) порівняно з плацебо (3,41 проти 3,90 на 100 пацієнто-років у групах ліраглутиду та плацебо відповідно), зменшивши ризик на 13%, відношення ризиків (HR) — 0,87, [0,78; 0,97] [95% ДІ]) (р = 0,005) (див. рисунок 4).

Рисунок 4. Графік Каплана — Маєра: час до першої події MACE — популяція FAS
Діти
Подвійно сліпе дослідження ефективності і безпеки застосування лікарського засобу Саксенда® порівняно з плацебо для зниження маси тіла у педіатричних пацієнтів віком від 12 років з ожирінням показало, що лікарський засіб Саксенда® є більш ефективним, ніж плацебо, для зниження маси тіла (яке оцінювалось за показником стандартного відхилення ІМТ) через 56 тижнів лікування (табл. 6).
Серед пацієнтів, які приймали ліраглутид, більший відсоток пацієнтів досяг зниження ІМТ на ≥ 5% та ≥ 10%, ніж серед пацієнтів, які приймали плацебо. В середньому зниження ІМТ та маси тіла також було більшим у групі пацієнтів, які приймали ліраглутид (табл. 6). Після 26 тижнів періоду подальшого спостереження без застосування досліджуваного лікарського засобу спостерігалося відновлення маси тіла у пацієнтів, які приймали ліраглутид, порівняно з тими, хто приймав плацебо (табл. 6).
Таблиця 6
Дослідження 4180: зміни від вихідного рівня маси тіла та ІМТ на 56-му тижні та зміна за показником стандартного відхилення ІМТ (ПСВ ІМТ) з 56-го тижня до 82-го тижня
|
|
Саксенда®
(N = 125)
|
Плацебо
(N = 126)
|
Саксенда® порівняно з плацебо
|
|
ПСВ ІМТ
|
|
|
|
|
Вихідний показник, ПСВ ІМТ (СВ)
|
3,14 (0,65)
|
3,20 (0,77)
|
|
|
Середня зміна на 56-му тижні
(95 % ДІ)
|
-0,23
|
-0,00
|
-0,22* (-0,37; -0,08)
|
|
56-й тиждень, ПСВ ІМТ (СВ)
|
2,88 (0,94)
|
3,14 (0,98)
|
|
|
Середня зміна з 56-го тижня до 82-го тижня, ПСВ ІМТ (95 % ДІ)
|
0,22
|
0,07
|
0,15** (0,07; 0,23)
|
|
Маса тіла
|
|
|
|
|
Вихідний показник, кг (СВ)
|
99,3 (19,7)
|
102,2 (21,6)
|
-
|
|
Середня зміна на 56-му тижні, % (95 % ДІ)
|
-2,65
|
2,37
|
-5,01** (-7,63; -2,39)
|
|
Середня зміна на 56-му тижні, кг (95 % ДІ)
|
-2,26
|
2,25
|
-4,50** (-7,17; -1,84)
|
|
ІМТ
|
|
|
|
|
Вихідний показник, кг/м2 (СВ)
|
35,3 (5,1)
|
35,8 (5,7)
|
-
|
|
Середня зміна на 56-му тижні, кг/м2 (95 % ДІ)
|
-1,39
|
0,19
|
-1,58** (-2,47; -0,69)
|
|
Відсоток пацієнтів зі зниженням на ≥ 5 % вихідного ІМТ на 56-му тижні, % (95 % ДІ)
|
43,25
|
18,73
|
3,31** (1,78; 6,16)
|
|
Відсоток пацієнтів зі зниженням на ≥ 10 % вихідного ІМТ на
56-му тижні, % (95 % ДІ)
|
26,08
|
8,11
|
4,00** (1,81; 8,83)
|
Повна вибірка пацієнтів. Для ПСВ ІМТ, маси тіла та ІМТ вихідні показники є середніми величинами, зміни від вихідного рівня на 56-му тижні є розрахунковими середніми величинами (методом найменших квадратів), а відмінності в лікуванні на 56-му тижні — це розрахункові різниці між методами лікування. Для ПСВ ІМТ показники на 56-му тижні є середніми величинами, зміни з 56-го тижня до 82-го тижня є розрахунковими величинами (методом найменших квадратів), а відмінності в лікуванні на 82-му тижні — це розрахункові різниці між методами лікування. Для частки пацієнтів, які втратили ≥ 5% / ≥ 10% вихідного ІМТ, також наведено розрахункове співвідношення шансів. Пропущені спостереження інтерпольовані з групи пацієнтів, які приймали плацебо, на основі переходу до референційного підходу множинної (x100) підстановки даних.
* p < 0,01, ** p < 0,001. ДІ — довірчий інтервал. СВ — стандартне відхилення.
За даними переносимості лікарського засобу, 103 пацієнти (82,4%) збільшили дозу до 3,0 мг та продовжували лікування цією дозою, 11 пацієнтів (8,8%) збільшили дозу до 2,4 мг і залишилися на цій дозі, 4 пацієнти (3,2%) збільшили дозу до 1,8 мг та продовжували лікування цією дозою, 4 пацієнти (3,2%) збільшили дозу до 1,2 мг та залишилися на лікуванні цією дозою, і 3 пацієнти (2,4%) приймали дозу 0,6 мг.
Через 56 тижнів лікування не було зареєстровано жодного впливу на ріст чи статеве дозрівання пацієнтів.
Дослідження з 16-тижневим подвійно сліпим періодом і 36-тижневим відкритим періодом було проведено для оцінки ефективності та безпеки застосування лікарського засобу Саксенда® педіатричним пацієнтам із синдромом Прадера — Віллі та ожирінням. Дослідження включало 32 пацієнти віком від 12 до 18 років (частина А) та 24 пацієнти віком від 6 до 12 років (частина В). Пацієнти були рандомізовані в співвідношенні 2:1 для прийому лікарського засобу Саксенда® або плацебо. Пацієнтам з масою тіла менше 45 кг почали збільшувати дозу на меншу величину (0,3 мг замість 0,6 мг) та збільшували до максимальної дози 2,4 мг.
Розрахункова різниця в лікуванні за середніми значеннями ПСВ ІМТ на 16 тижні (частина A: -0,20 проти -0,13, частина B: -0,50 проти -0,44) і на 52 тижні (частина A: -0,31 проти -0,17, частина B: -0,73 проти — 0,67) була подібною при застосуванні лікарського засобу Саксенда® та плацебо.
Жодних додаткових проблем щодо безпеки під час дослідження не було помічено.
Європейське агентство з лікарських засобів відклало зобов’язання подавати результати досліджень застосування лікарського засобу Саксенда® в одній або декількох підгрупах педіатричних пацієнтів при лікуванні ожиріння та при лікуванні синдрому Прадера-Віллі (див. розділ «Спосіб застосування та дози»).
Фармакокінетика
Абсорбція
Абсорбція ліраглутиду після підшкірного введення відбувається повільно, максимальна концентрація досягається приблизно через 11 годин після введення. У пацієнтів з ожирінням (ІМТ 30–40 кг/м2) після введення 3 мг ліраглутиду його середня рівноважна концентрація (AUCt/24) досягала приблизно 31 нмоль/л. Експозиція ліраглутиду збільшувалася пропорційно дозі. Абсолютна біодоступність ліраглутиду після підшкірного введення становить приблизно 55%.
Розподіл
Середній видимий об’єм розподілу після підшкірного введення становить 20–25 л (для людини з масою тіла приблизно 100 кг). Ліраглутид значною мірою зв’язується з білками плазми крові (> 98%).
Метаболізм
Протягом 24 годин після введення разової дози [3H]-ліраглутиду здоровим добровольцям основним компонентом у плазмі крові був незмінений ліраглутид. У плазмі крові були виявлені в незначній кількості два метаболіти (≤ 9% і ≤ 5% від загального рівня радіоактивності у плазмі крові).
Виведення
Ліраглутид ендогенно метаболізується, як і всі великі білки, не маючи специфічного органа як основного шляху елімінації. Після введення дози [3H]-ліраглутиду в сечі і калі не було виявлено незміненого ліраглутиду. Тільки невелика частка введеної радіоактивності у вигляді метаболітів ліраглутиду виводилась нирками та через кишечник (6% і 5% відповідно). Радіоактивні речовини виводяться нирками або через кишечник в основному протягом перших 6–8 діб у вигляді трьох метаболітів.
Після одноразового підшкірного введення ліраглутиду середнє значення кліренсу становить приблизно 0,9–1,4 л/годину, період напіввиведення — приблизно 13 годин.
Особливі групи пацієнтів
Пацієнти літнього віку
На підставі даних фармакокінетичного аналізу групи пацієнтів віком від 18 до 82 років з надмірною масою тіла чи ожирінням був зроблений висновок, що вік не має клінічно значущого впливу на фармакокінетику ліраглутиду. Тому немає необхідності в коригуванні дози залежно від віку пацієнта.
Стать
Дані фармакокінетичного аналізу показали, що у жінок спостерігається на 24% нижчий кліренс ліраглутиду порівняно з чоловіками. На підставі цих даних можна зробити висновок, що корекція дози залежно від статі пацієнта не потрібна.
Етнічне походження
На підставі даних фармакокінетичного аналізу групи пацієнтів європеоїдної, монголоїдної і негроїдної рас та латиноамериканців з надмірною масою тіла чи ожирінням був зроблений висновок, що етнічне походження пацієнта не виявляє будь-якого істотного клінічного впливу на фармакокінетику ліраглутиду.
Маса тіла
Експозиція ліраглутиду зменшується зі збільшенням початкової маси тіла. Як показали дослідження, добова доза ліраглутиду 3,0 мг забезпечує нормальний системний вплив на організм пацієнта з масою тіла 60–234 кг. Експозиція ліраглутиду у пацієнтів з масою тіла більше 234 кг не вивчалась.
Порушення функції печінки
Фармакокінетику ліраглутиду досліджували у пацієнтів із різним ступенем порушень функції печінки при застосуванні одноразової дози (0,75 мг). Було показано, що у пацієнтів з легкими і помірними порушеннями функції печінки експозиція ліраглутиду знижувалася на 13–23% порівняно зі здоровими добровольцями. У пацієнтів з тяжкими порушеннями функції печінки (> 9 балів за класифікацією Чайлда — П’ю) експозиція була значно нижча (на 44%).
Порушення функції нирок
Експозиція ліраглутиду була знижена у пацієнтів з порушеннями функції нирок порівняно з особами з нормальною функцією нирок у процесі дослідження із застосуванням одноразової дози (0,75 мг). У пацієнтів з легкими порушеннями (кліренс креатиніну 50–80 мл/хв) експозиція знижувалася на 33%, з порушеннями помірної тяжкості (кліренс креатиніну 30–50 мл/хв) — на 14%, з тяжкими порушеннями (кліренс креатиніну < 30 мл/хв) — на 27%, а на кінцевих стадіях захворювань нирок, що вимагають проведення діалізу, — на 26%.
Діти
Фармакокінетичні властивості ліраглутиду в дозі 3,0 мг було оцінено в ході клінічних досліджень за участю педіатричних пацієнтів віком ≥ 12 років з ожирінням (134 пацієнти з масою тіла 62–178 кг).
Експозиція ліраглутиду у дітей віком ≥ 12 років була подібною до експозиції у дорослих з ожирінням.
Фармакокінетичні властивості також оцінювали у ході клініко-фармакологічного дослідження за участю педіатричних пацієнтів віком 7–11 років з ожирінням (13 пацієнтів, маса тіла 54–87 кг) .
Було виявлено порівнянну експозицію при введенні 3,0 мг ліраглутиду у дорослих, дітей віком ≥ 12 років та дітей віком 7–11 років після корекції за масою тіла.
Доклінічні дані з безпеки
Доклінічні дані, що базуються на дослідженнях з фармакологічної безпеки, токсичності повторних доз та генотоксичності, не виявили жодного ризику для людини.
У процесі дворічних досліджень канцерогенності у щурів та мишей були виявлені пухлини С-клітин щитоподібної залози, що не призводили до летального результату. Нетоксична доза для щурів не була встановлена. У мавп, що отримували лікування протягом 20 місяців, таких пухлин не виявлено. Пухлини у гризунів обумовлені негенотоксичним специфічним ГПП-1-рецепторопосередкованим механізмом, до якого частково чутливі гризуни. Значущість цього механізму для людини достатньо низька, але не може бути повністю знехтувана. Розвитку інших пухлин не було виявлено. У процесі досліджень на тваринах не було виявлено прямого шкідливого впливу на фертильність, проте при введенні найвищих доз відзначалося незначне підвищення ранньої ембріональної летальності. Введення ліраглутиду в період середини вагітності спричиняло зниження маси тіла самки, уповільнення росту плода з нез’ясованим впливом на розвиток ребер у щурів і скелета у кролів. При введенні ліраглутиду відзначено уповільнення росту новонароджених щурів, що зберігається в період відлучення від годування молоком у групі прийому високої дози. Невідомо, чи уповільнення росту новонароджених щурів обумовлене зниженням споживання ними молока в результаті прямого впливу ГПП-1 чи зменшенням молока у матері, що обумовлено зниженням калорійності споживаної їжі.
У ювенільних щурів застосування ліраглутиду призводило до затримки статевого дозрівання як самців, так і самок при клінічно значущих концентраціях препарату в плазмі крові. Такі затримки не впливали на фертильність і репродуктивну здатність самок та самців чи на здатність самок виношувати вагітність.
Клінічні характеристики
Показання
Дорослі
Лікарський засіб Саксенда® застосовують для зменшення маси тіла як доповнення до дієти зі зниженою калорійністю та збільшеною фізичною активністю у дорослих пацієнтів з початковим індексом маси тіла (ІМТ) більше 30 кг/м2 (ожиріння) або від 27 до 30 кг/м2 (надмірна маса тіла) за наявності хоча б одного супутнього захворювання, пов’язаного з масою тіла, такого як дисглікемія (переддіабет або цукровий діабет 2 типу), гіпертензія, дисліпідемія або обструктивне апное сну.
Якщо через 12 тижнів після прийому добової дози 3,0 мг пацієнт не втратив щонайменше 5% від початкової маси тіла, застосування лікарського засобу Саксенда® слід припинити.
Діти віком ≥ 12 років
Лікарський засіб Саксенда® можна застосовувати як доповнення до здорового харчування та збільшеної фізичної активності для корекції маси тіла у дітей віком від 12 років із:
- ожирінням (ІМТ, що відповідає ≥ 30 кг/м2 для дорослих пацієнтів за міжнародними гранично допустимими значеннями)* та
- масою тіла більш ніж 60 кг.
Лікування препаратом Саксенда® слід припинити і переглянути, якщо пацієнт не втратив щонайменше 4% свого ІМТ або ІМТ за Z-показником через 12 тижнів застосування препарату в дозі 3,0 мг/добу чи в максимальній переносимій дозі.
* Гранично допустимі значення ІМТ, встановлені Міжнародною робочою групою з вивчення ожиріння, для пацієнтів віком 12–18 років із ожирінням залежно від статі пацієнта (див. табл. 7).
Таблиця 7
Гранично допустимі значення ІМТ, встановлені Міжнародною робочою групою з вивчення ожиріння, для пацієнтів віком 12–18 років із ожирінням залежно від статі пацієнта
|
Вік
(років)
|
ІМТ, який відповідає 30 кг/м2 для дорослих пацієнтів за міжнародними гранично допустимими значеннями
|
|
Хлопці
|
Дівчата
|
|
12
|
26,02
|
26,67
|
|
12,5
|
26,43
|
27,24
|
|
13
|
26,84
|
27,76
|
|
13,5
|
27,25
|
28,20
|
|
14
|
27,63
|
28,57
|
|
14,5
|
27,98
|
28,87
|
|
15
|
28,30
|
29,11
|
|
15,5
|
28,60
|
29,29
|
|
16
|
28,88
|
29,43
|
|
16,5
|
29,14
|
29,56
|
|
17
|
29,41
|
29,69
|
|
17,5
|
29,70
|
29,84
|
|
18
|
30,00
|
30,00
|
Протипоказання
Підвищена чутливість до діючої речовини або до інших компонентів лікарського засобу.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Іn vitro ліраглутид продемонстрував дуже низький потенціал впливу на фармакокінетику інших активних субстанцій, обмін яких пов’язаний із цитохромом Р450, а також зв’язування з білками плазми крові.
Ліраглутид спричинює незначну затримку випорожнення шлунка, що може вплинути на всмоктування пероральних препаратів, які застосовуються одночасно. Дослідження щодо взаємодії не показали будь-якого клінічно значущого уповільнення всмоктування, тому корекція дози не потрібна.
Дослідження взаємодії проводили при застосуванні ліраглутиду в дозі 1,8 мг. Вплив на швидкість випорожнення шлунка був еквівалентним у ліраглутиду в дозі 1,8 мг та 3,0 мг (парацетамол AUC0–300 хв). Зареєстрований щонайменше один епізод виникнення гострої діареї у деяких пацієнтів, які отримували лікарський засіб Саксенда®. Діарея може порушувати всмоктування пероральних лікарських засобів, що приймаються одночасно.
Варфарин та інші похідні кумарину
Досліджень лікарської взаємодії не проводили. Не можна виключити клінічно значущу взаємодію з активною субстанцією, що має низьку розчинність або вузький терапевтичний індекс, такою як варфарин. На початку лікування ліраглутидом у пацієнтів, які одержують варфарин або інші похідні кумарину, рекомендується частіше проводити контроль міжнародного нормалізованого співвідношення (МНС).
Парацетамол
Ліраглутид не змінював загальну експозицію парацетамолу після введення одноразової дози 1000 мг. Максимальна концентрація парацетамолу (Cmax) знижувалася на 31%, а медіана часу досягнення максимальної концентрації (tmax) збільшувалася до 15 хвилин. При одночасному застосуванні парацетамолу корекція дози не потрібна.
Аторвастатин
Ліраглутид не змінював загальну експозицію аторвастатину до клінічно значущого рівня після одноразового його введення в дозі 40 мг. У зв’язку з цим при одночасному застосуванні з ліраглутидом корекція дози аторвастатину не потрібна. При одночасному введенні з ліраглутидом Cmax аторвастатину знижувалася на 38%, а медіана tmax збільшувалася з 1 години до 3 годин.
Гризеофульвін
Ліраглутид не змінював загальної експозиції гризеофульвіну після одноразового його введення в дозі 500 мг. Cmax гризеофульвіну зростала на 37%, тоді як медіана tmax не змінювалася. Коригування дози гризеофульвіну та інших низькорозчинних сполук з високою проникністю не потрібне.
Дигоксин
Після одноразового введення 1 мг дигоксину у поєднанні з ліраглутидом відмічено зменшення площі під кривою «концентрація-час» (AUC) дигоксину на 16%; Cmax знижувалася на 31%; медіана tmax дигоксину збільшувалася з 1 години до 1,5 години. З огляду на зазначені результати корекція дози дигоксину не потрібна.
Лізиноприл
Після одноразового застосування 20 мг лізиноприлу з ліраглутидом відмічено зменшення AUC лізиноприлу на 15%, Cmax знижувалася на 27%, а медіана tmax лізиноприлу збільшувалася з 6 до 8 годин. З огляду на зазначені результати корекція дози лізиноприлу не потрібна.
Пероральні контрацептиви
При одночасному застосуванні разової дози пероральних контрацептивів ліраглутид знижував Cmax етинілестрадіолу або левоноргестрелу на 12% і 13% відповідно, а tmax збільшувався на 1,5 години. Це не мало клінічно значущого впливу на загальну експозицію етинілестрадіолу або левоноргестрелу, що дає підставу вважати, що одночасний прийом їх з ліраглутидом не вплине на контрацептивний ефект етинілестрадіолу та левоноргестрелу.
Діти
Дослідження лікарської взаємодії проводились лише за участю дорослих пацієнтів.
Особливості застосування
Відстежування
З метою покращення відстежування біологічних лікарських засобів слід зазначати у медичній документації пацієнта назву та номер серії введеного препарату.
Аспірація в поєднанні із загальною анестезією або глибокою седацією
У пацієнтів, які отримували агоністи рецепторів ГПП-1, спостерігалися випадки легеневої аспірації під час їх перебування під загальною анестезією або у стані глибокої седації. Тому перед виконанням процедури із використанням загальної анестезії або глибокої седації слід враховувати підвищений ризик наявності залишкового вмісту шлунка внаслідок затримки випорожнення шлунка (див. розділ «Побічні реакції»).
Серцева недостатність
Немає клінічного досвіду лікування пацієнтів із застійною серцевою недостатністю IV класу за класифікацією Нью-Йоркської асоціації кардіологів (NYHA), тому ліраглутид не рекомендовано застосовувати цим пацієнтам.
Особливі групи пацієнтів
Безпека та ефективність застосування ліраглутиду для корекції маси тіла не встановлені для пацієнтів:
- віком ≥ 75 років;
- які застосовують інші лікарські засоби для корекції маси тіла;
- із вторинним ожирінням, викликаним ендокринологічними розладами чи розладами, пов’язаними з харчуванням, або в результаті застосування лікарських засобів, що можуть спричинити збільшення маси тіла;
- з тяжким порушенням функції нирок;
- з тяжким порушенням функції печінки.
Не рекомендується застосовувати лікарський засіб Саксенда® цим групам пацієнтів (див. розділ «Спосіб застосування та дози»).
Оскільки дослідження щодо застосування ліраглутиду для корекції маси тіла пацієнтам з легкими або помірними порушеннями функції печінки відсутні, його слід з обережністю застосовувати цій групі пацієнтів (див. розділи «Фармакокінетика» та «Спосіб застосування та дози»).
Досвід застосування ліраглутиду пацієнтам із запальними захворюваннями кишечнику і діабетичним гастропарезом обмежений. Застосування ліраглутиду цим пацієнтам не рекомендовано, оскільки воно супроводжується тимчасовими побічними реакціями з боку шлунково-кишкового тракту, в т. ч. нудотою, блюванням і діареєю.
Панкреатит
Спостерігались випадки гострого панкреатиту при застосуванні аналогів рецептора ГПП-1.
Пацієнтів слід проінформувати про характерні симптоми гострого панкреатиту. При підозрі на панкреатит слід відмінити лікування ліраглутидом. Якщо підтверджується гострий панкреатит, повторне застосування ліраглутиду не рекомендоване.
Жовчнокам’яна хвороба та холецистит
У клінічних випробуваннях у пацієнтів, які застосовували ліраглутид для зменшення маси тіла, спостерігалася більша частота виникнення жовчнокам’яної хвороби та холециститу порівняно з пацієнтами, які отримували плацебо. Той факт, що швидка втрата маси тіла може збільшити ризик розвитку жовчнокам’яної хвороби і, отже, холециститу, лише частково пояснює більш високу частоту виникнення цих захворювань при застосуванні ліраглутиду. Жовчнокам’яна хвороба та холецистит можуть призвести до госпіталізації та холецистектомії. Пацієнтів слід поінформувати про характерні симптоми жовчнокам’яної хвороби та холециститу.
Захворювання щитоподібної залози
У процесі клінічних досліджень цукрового діабету 2 типу відмічені побічні реакції з боку щитоподібної залози, такі як зоб, особливо у пацієнтів з уже наявними захворюваннями щитоподібної залози. Тому ліраглутид слід з обережністю застосовувати таким пацієнтам.
Частота серцевих скорочень
Під час клінічних досліджень ліраглутиду спостерігалося збільшення частоти серцевих скорочень (див. розділ «Фармакодинаміка»). Частоту серцевих скорочень слід контролювати через рівні проміжки часу відповідно до звичайної клінічної практики. Пацієнтів слід проінформувати про симптоми збільшення частоти серцевих скорочень (підвищене серцебиття або відчуття підвищеного серцебиття в спокої). Пацієнтам, у яких спостерігається клінічно значуще стійке збільшення частоти серцевих скорочень у спокої, лікування ліраглутидом слід припинити.
Зневоднення
У пацієнтів, які застосовували агоністи рецепторів ГПП-1, спостерігалися симптоми зневоднення, в тому числі порушення функції нирок та гостра ниркова недостатність. Пацієнтів, яким призначено ліраглутид, потрібно проінформувати про можливість зневоднення організму внаслідок розладів травної системи та про необхідність вжиття запобіжних заходів щодо зневоднення.
Гіпоглікемія у пацієнтів із цукровим діабетом 2 типу
У пацієнтів із цукровим діабетом 2 типу, які отримують ліраглутид одночасно з інсуліном та/або сульфонілсечовиною, може бути підвищений ризик виникнення гіпоглікемії. Ризик виникнення гіпоглікемії може бути знижений за допомогою зменшення дози інсуліну та/або сульфонілсечовини.
Діти
У дітей віком ≥ 12 років, які отримували лікування ліраглутидом, повідомлялося про епізоди клінічно значущої гіпоглікемії. Пацієнтів слід проінформувати про характерні симптоми гіпоглікемії та відповідні заходи.
Гіперглікемія у пацієнтів із цукровим діабетом 2 типу, які отримують інсулін
Пацієнтам із цукровим діабетом 2 типу лікарський засіб Саксенда® не можна застосовувати як заміну інсуліну. Повідомлялося про розвиток діабетичного кетоацидозу у інсулінозалежних пацієнтів у разі швидкого припинення застосування або зменшення дози інсуліну (див. розділ «Спосіб застосування та дози»).
Допоміжні речовини
Саксенда® містить менше ніж 1 ммоль натрію (23 мг), тому лікарський засіб можна вважати таким, що не містить натрію.
Застосування у період вагітності або годування груддю
Вагітність
Адекватні дані щодо застосування ліраглутиду вагітним жінкам відсутні. Дослідження на тваринах показали репродуктивну токсичність (див. розділ «Фармакологічні властивості», підрозділ «Доклінічні дані з безпеки»). Потенційний ризик для людей невідомий.
Ліраглутид не слід застосовувати під час вагітності. Якщо пацієнтка хоче завагітніти або вагітна, то прийом ліраглутиду необхідно відмінити.
Період годування груддю
Невідомо, чи екскретується ліраглутид у грудне молоко людини. Дослідження на тваринах показали, що в молоко потрапляє незначна кількість ліраглутиду і його близькоспоріднених структурних метаболітів. Доклінічні дослідження виявили пов’язане із застосуванням препарату зменшення темпів зростання новонароджених щуренят (див. розділ «Фармакологічні властивості», підрозділ «Доклінічні дані з безпеки»). У зв’язку з недостатнім досвідом застосування препарату у період годування груддю не слід застосовувати його в цей період.
Фертильність
Окрім незначного зменшення кількості живих імплантованих ембріонів, дослідження на тваринах не виявили шкідливого впливу препарату на репродуктивну здатність (див. розділ «Фармакологічні властивості», підрозділ «Доклінічні дані з безпеки»).
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
Лікарський засіб Саксенда® не впливає або має незначний вплив на здатність керувати транспортними засобами й іншими механізмами. Однак може виникати запаморочення, здебільшого протягом перших 3 місяців лікування лікарським засобом Саксенда®. Слід з обережністю керувати автотранспортом або іншими механізмами при виникненні запаморочення.
Спосіб застосування та дози
Дозування
Дорослі
Початкова доза становить 0,6 мг на добу. Для поліпшення переносимості з боку шлунково-кишкового тракту дозу слід підвищувати щотижня на 0,6 мг до досягнення добової дози 3,0 мг (див. таблицю 8).
У разі поганої переносимості наступної підвищеної дози протягом двох послідовних тижнів слід розглянути питання про припинення лікування. Добова доза вище 3,0 мг не рекомендується.
Таблиця 8
Графік підвищення дози
| |
Доза, мг
|
Тижні
|
|
Збільшення дози протягом
4 тижнів
|
0,6
|
1
|
|
1,2
|
1
|
|
1,8
|
1
|
|
2,4
|
1
|
|
Підтримуюча доза
|
3,0 мг
|
Діти віком ≥ 12 років
Для дітей віком від 12 років слід застосовувати таку ж схему підвищення дози, як і для дорослих (див. таблицю 8). Дозу слід збільшувати до досягнення 3,0 мг (підтримуюча доза) або максимальної переносимої дози. Добові дози вище 3,0 мг не рекомендуються.
Пропущена доза
Якщо з часу звичайного проведення ін’єкції пройшло не більше 12 годин, пацієнт повинен прийняти пропущену дозу якнайшвидше. Якщо до наступного введення залишається менше 12 годин, пацієнт не повинен приймати пропущену дозу, а продовжувати режим прийому 1 раз на добу — прийняти наступну заплановану дозу препарату. Не слід приймати додаткову дозу чи збільшувати дозу для компенсації пропущеної ін’єкції.
Пацієнти з цукровим діабетом 2 типу
Лікарський засіб Саксенда® не слід застосовувати в комбінації з іншими агоністами рецепторів ГПП-1. Для зниження ризику розвитку гіпоглікемії на початку застосування лікарського засобу Саксенда® слід розглянути можливість зменшення дози одночасно застосовуваних інсуліну або стимуляторів секреції інсуліну (наприклад сульфонілсечовини). Необхідний самоконтроль рівня глюкози в крові для коригування дози інсуліну чи стимуляторів секреції інсуліну (див. розділ «Особливості застосування»).
Особливі групи пацієнтів
Пацієнти літнього віку (≥ 65 років)
Корекція дози у зв’язку з віком пацієнта не потрібна. Досвід застосування препарату пацієнтам віком ≥ 75 років обмежений, тому не рекомендовано застосовувати його цій категорії пацієнтів (див. розділи «Фармакокінетика» та «Особливості застосування»).
Пацієнти з порушеннями функції нирок
Корекція дози не потрібна пацієнтам з легким або помірним ступенем порушення функції нирок (кліренс креатиніну ≥ 30 мл/хв). Не рекомендується застосовувати лікарський засіб Саксенда® пацієнтам з тяжким порушенням функції нирок (кліренс креатиніну < 30 мл/хв), включаючи пацієнтів із термінальною стадією порушення функції нирок (див. розділи «Фармакокінетика», «Особливості застосування» та «Побічні реакції»).
Пацієнти з порушеннями функції печінки
Не рекомендується коригування дози пацієнтам з легким або помірним ступенем порушення функції печінки. Застосування лікарського засобу Саксенда® не рекомендується пацієнтам з тяжким порушенням функції печінки, а пацієнтам із легким або помірним порушенням функції печінки препарат слід застосовувати з обережністю (див. розділи «Фармакокінетика» та «Особливості застосування»).
Спосіб введення
Лікарський засіб Саксенда® призначений тільки для підшкірного введення. Його не можна вводити внутрішньовенно або внутрішньом’язово.
Препарат вводять підшкірно 1 раз на добу у будь-який час незалежно від вживання їжі. Його можна вводити підшкірно в ділянку передньої черевної стінки, стегна або плеча. Місце і час введення можна змінювати без корекції дози, проте бажано вводити приблизно в один і той же найбільш зручний час.
Нижче наводиться інструкція з використання шприц-ручки для введення лікарського засобу.
З метою зменшення ризику утворення відкладень амілоїду при ін’єкції необхідно завжди змінювати місце ін’єкції (див. розділ «Побічні реакції»).
Інструкція з використання шприц-ручки Саксенда®
Перед використанням шприц-ручки Саксенда® необхідно уважно прочитати цю інструкцію.
Для детальної інформації див. інструкцію для медичного застосування. Не слід користуватися шприц-ручкою без отримання належної інформації щодо її використання від лікаря чи медсестри.
Застосування препарату необхідно почати з перевірки шприц-ручки, щоб бути впевненим, що вона містить саме лікарський засіб Саксенда®, 6 мг/мл. Потім потрібно подивитися рисунки нижче, щоб отримати інформацію про різні частини шприц-ручки та голки.
Незрячим пацієнтам та пацієнтам з поганим зором не можна користуватися шприц-ручкою без сторонньої допомоги. Допомагати має людина з хорошим зором, яка може побачити лічильник дози на шприц-ручці Саксенда® та яка вміє користуватися цією шприц-ручкою.
Розчин для ін’єкцій у попередньо заповненій шприц-ручці. Шприц-ручка Саксенда® є попередньо заповненою. Вона містить 18 мг ліраглутиду, що дає змогу ввести дози 0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг та 3,0 мг. Шприц-ручка призначена для використання з одноразовими голками НовоФайн® або НовоТвіст® довжиною до 8 мм і товщиною 32G. Голки до комплекту упаковки не входять.
· Важлива інформація
Необхідно звернути особливу увагу на цю позначку, оскільки вона є важливою для безпечного користування шприц-ручкою.
Шприц-ручка Саксенда® та голка (приклад)

|
1. Підготовка шприц-ручки з новою голкою для використання
• Перевірте назву та кольорову етикетку
Вашої шприц-ручки, щоб бути впевненим, що вона містить лікарський засіб Саксенда®. Це особливо важливо у разі, коли Ви застосовуєте різні ін’єкційні лікарські засоби. Помилкове застосування лікарського засобу може бути шкідливим для Вашого здоров’я.
• Зніміть ковпачок шприц-ручки
|

|
- Переконайтеся, що розчин у шприц-ручці прозорий та безбарвний. Подивіться у вікно шкали картриджа. Якщо препарат мутний, шприц-ручку використовувати заборонено.
|

|
- Візьміть нову одноразову голку та видаліть з неї захисну мембрану.
|

|
- Переконайтеся, що голка прикріплена правильно.
• Нагвинтіть голку на шприц-ручку та поверніть її, щоб голка щільно трималась на шприц-ручці
|

|
|
• Голка закрита двома ковпачками. Ви повинні зняти обидва ковпачки
Якщо ви забудете зняти обидва ковпачки, ви не зробите ін’єкцію розчину.
• Зніміть зовнішній ковпачок голки та збережіть його
Він знадобиться після завершення ін’єкції для безпечного зняття голки.
|

|
- Зніміть внутрішній ковпачок голки та викиньте його. Якщо Ви спробуєте надіти внутрішній ковпачок знову на голку, то можете поранитись. На кінці голки може з’явитися крапля розчину. Це нормальне явище, проте необхідно перевірити надходження препарату при використанні нової шприц-ручки вперше.
Не приєднуйте нову голку
доти, доки не будете готові зробити ін’єкцію.
- Для кожної ін’єкції завжди використовуйте нову голку. Це зменшить ризик закупорки голки, зараження, потрапляння інфекції та введення неправильної дози препарату.
· Ніколи не використовуйте голку, якщо вона погнута чи пошкоджена
|

|
|
2. Перевірка потоку розчину з кожної нової шприц-ручки
- Якщо Ви вже використовували цю шприц-ручку, переходьте до пункту 3 «Виставлення дози». Перевіряйте потік розчину лише перед першим застосуванням нової шприц-ручки.
• Повертайте селектор дози до символу перевірки потоку
(  ) прямо після «0». Переконайтеся, що символ перевірки потоку збігається з покажчиком дози. ) прямо після «0». Переконайтеся, що символ перевірки потоку збігається з покажчиком дози.
|

|
- Тримайте шприц-ручку голкою вгору.
Натисніть і потримайте кнопку дози, поки лічильник дози не повернеться до «0». Значення «0» повинно відповідати покажчику дози. На кінчику голки повинна з’явитися крапля розчину.
На кінчику голки може залишитися невелика крапля, але вона не буде вводитися.
Якщо крапля не з’явилася, поверніться до пункту 2 «Перевірка потоку розчину з нової шприц-ручки» до 6 разів. Якщо краплі все ще немає, змініть голку і повторіть дії, описані у пункті 2 «Перевірка потоку розчину з нової шприц-ручки» ще раз.
Якщо крапля все-таки не з’явилася, утилізуйте ручку і використовуйте нову.
- Потрібно завжди переконуватись, що крапля з’являється на кінчику голки, перш ніж вперше використовувати нову ручку. Це гарантує, що розчин буде введений.
Якщо крапля не з’явиться, не використовуйте шприц-ручку, навіть якщо лічильник дози змінює значення. Це може вказувати на заблоковану або пошкоджену голку.
Якщо Ви не перевірите потік перед першою ін’єкцією нової ручки, то можете не отримати необхідну дозу розчину для забезпечення бажаної дії лікарського засобу Саксенда®.
|

|
|
3. Виставлення дози
- Повертайте селектор дози, поки лічильник дози не покаже необхідну дозу (0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг або 3,0 мг).
Якщо Ви вибрали неправильну дозу, можна повернути селектор дози вперед або назад до правильної дози.
Шприц-ручка вміщує максимум 3,0 мг препарату.
За допомогою селектора можна змінити дозу. Тільки лічильник дози та показник дози покажуть, яку кількість міліграмів Ви вибираєте для ін’єкції.
Ви можете вибрати до 3,0 мг лікарського засобу на дозу. Якщо шприц-ручка містить менше 3,0 мг, лічильник дози зупиняється до значення 3,0 мг.
Чути різне клацання селектора дози при обертанні вперед, назад або при виставленні залишкової кількості розчину. Не рахуйте клацання.
 Завжди використовуйте лічильник дози та покажчик дози, щоб побачити, скільки міліграмів Ви вибрали перед введенням цього препарату. Завжди використовуйте лічильник дози та покажчик дози, щоб побачити, скільки міліграмів Ви вибрали перед введенням цього препарату.
Не рахуйте кількість клацань шприц-ручки.
Не використовуйте шкалу картриджа. Вона показує приблизну кількість розчину, яка залишилась у шприц-ручці.
За допомогою селектора дози повинні бути виставлені лише дози 0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг або 3,0 мг
Цифри на дисплеї повинні точно збігатися з покажчиком дози, щоб забезпечити правильну дозу для введення.
|

|
|
Залишок розчину в шприц-ручці
- Шкала шприц-ручки показує приблизно, скільки розчину залишилось у шприц-ручці.
|

|
- Щоб точно побачити, скільки розчину залишилось, скористайтеся лічильником дози:
Повертайте селектор дози вліво, поки лічильник дози не зупиниться.
Якщо він показує 3,0, у шприц-ручці залишилося щонайменше 3,0 мг. Якщо лічильник дози зупиняється до 3,0 мг, значить у шприц-ручці не вистачає розчину для повної дози 3,0 мг.
Якщо Вам потрібна вища доза, ніж та, що залишилася у Вашій шприц-ручці
Ви можете розділити свою дозу між Вашою поточною шприц-ручкою та новою шприц-ручкою тільки за умови, що Вас проінструктував лікар або медсестра. Використовуйте калькулятор, щоб спланувати дози згідно з інструкціями лікаря або медсестри. Будьте дуже обережні, щоб правильно розрахувати дозу.
· Будьте дуже уважні, розраховуючи дозу
Якщо Ви не впевнені, що зможете розділити дозу за допомогою двох шприц-ручок, виберіть і введіть необхідну дозу новою шприц-ручкою.
|

|
|
4. Введення дози
- Введіть голку під шкіру так, як показав Вам лікар чи медсестра.
• Переконайтеся, що Ви бачите лічильник дози
Не закривайте лічильник пальцями. Це може перервати ін’єкцію.
|

|
|
• Натисніть і утримуйте пускову кнопку. Спостерігайте, поки лічильник дози не повернеться до «0»
«0» повинен збігатися з покажчиком дози. Потім Ви можете почути або відчути клацання.
- Продовжуйте натискати пускову кнопку, утримуючи голку під шкірою.
|

|
- Рахуйте повільно до 6, утримуючи натиснутою пускову кнопку.
- Якщо голку витягти раніше, Ви можете побачити потік розчину з кінчика голки. В такому випадку правильна доза не буде введена.
|

|
- Витягніть голку з-під шкіри. Після цього можна відпустити пускову кнопку.
Якщо крапля крові з’явилась у місці ін’єкції, легенько притисніть це місце.
Ви можете побачити краплю розчину на кінчику голки після введення. Це нормально і не впливає на об’єм введеної дози.
· Завжди дивіться на покажчик дози, щоб бачити, скільки міліграмів розчину Ви ввели
Тримайте натиснутою пускову кнопку, поки покажчик дози не покаже знову «0».
Як виявити заблоковану чи пошкоджену голку?
- Якщо «0» не відображається на лічильнику дози після постійного натискання пускової кнопки, можливо, Ви використали заблоковану або пошкоджену голку.
- У цьому випадку Ви не ввели потрібну кількість лікарського засобу, навіть незважаючи на те, що лічильник дози перемістився від раніше встановленої дози.
Як поводитися із заблокованою голкою?
Зніміть голку, як описано в пункті 5 «Після ін’єкції», і повторіть дії, описані в усіх пунктах, починаючи з пункту 1 «Підготовка шприц-ручки з новою голкою для використання».
Ніколи не торкайтеся лічильника під час введення лікарського засобу
Це може перервати ін’єкцію.
|

|
|
5. Після ін’єкції
- Завжди утилізуйте голку після кожної ін’єкції, щоб забезпечити зручність ін’єкції та запобігти використанню заблокованої голки. Якщо голка заблокована, Ви не зможете ввести лікарський засіб.
- Покладіть зовнішній ковпачок голки на пласку поверхню і надіньте зовнішній ковпачок на голку, не торкаючись голки та зовнішнього ковпачка голки
|

|
|
• Як тільки голка накривається, обережно повністю натисніть на зовнішній ковпачок голки.
- Обережно викрутіть голку та утилізуйте за інструкціями, отриманими від лікаря, медсестри, фармацевта, або відповідно до місцевих правил.
|

|
- Закривайте кришкою шприц-ручку після кожного використання, щоб захистити розчин від світла.
Коли ручка порожня, викиньте її без голки за інструкціями, отриманими від лікаря, медсестри, фармацевта, або відповідно до місцевих правил.
- Ніколи не намагайтеся надіти внутрішній ковпачок голки назад на голку. Ви можете поранитися голкою.
- Завжди виймайте голку з ручки після кожної ін’єкції. Це може запобігти заблокуванню голки, забрудненню, зараженню, протіканню розчину та неточному дозуванню.
|

|
|
• Важлива інформація
- Завжди тримайте шприц-ручку та голки в недоступному місці для інших людей, особливо дітей.
- Ніколи не передавайте Вашу особисту шприц-ручку чи голки іншим людям. Призначений лише для індивідуального використання.
- Люди, що доглядають, мають бути дуже обережними при роботі з використаними голками, щоб запобігти травмуванню голкою та перехресному зараженню.
|
|
|
Догляд за Вашою шприц-ручкою
- Не залишайте Вашу шприц-ручку в машині чи іншому місці, де може бути дуже жарко або занадто холодно.
- Не вводьте лікарський засіб Саксенда®, який був заморожений. У такому разі ви можете не отримати очікуваного лікувального ефекту від цього препарату.
- Запобігайте контакту шприц-ручки з пилом, брудом чи рідиною.
- Не мийте, не замочуйте та не змащуйте шприц-ручку. Її можна очистити за допомогою тканини, змоченої м’яким миючим засобом.
- Запобігайте падінню та ударам шприц-ручки об тверді поверхні. В разі падіння чи удару приєднайте нову голку і перевірте подачу розчину перед введенням.
- Не намагайтеся повторно наповнити шприц-ручку. Утилізуйте шприц-ручку після закінчення в ній розчину. Утилізуйте шприц-ручку через місяць після першого використання
- Не намагайтеся самостійно полагодити шприц-ручку або розібрати її на частини.
- Не зберігати шприц-ручку з приєднаною голкою
|
|
Діти
Для дітей віком від 12 років корекція дози не потрібна. Безпеку та ефективність застосування лікарського засобу Саксенда® дітям віком до 12 років не встановлено (див. розділ «Фармакодинаміка»).
Передозування
У клінічних дослідженнях та протягом постмаркетингового застосування лікарського засобу Саксенда® повідомлялося про випадки передозування до 72 мг (у 24 рази більше рекомендованої підтримуючої дози). Події, про які повідомлялося, включали сильну нудоту, сильне блювання та тяжку гіпоглікемію.
У разі передозування слід розпочати підтримуюче лікування відповідно до наявних у пацієнта клінічних ознак і симптомів. Необхідно спостерігати за станом пацієнта щодо клінічних ознак зневоднення та контролювати рівень глюкози в крові.
Побічні реакції
Резюме профілю безпеки
Безпека застосування лікарського засобу Саксенда® була оцінена у 5 подвійно сліпих рандомізованих плацебо-контрольованих клінічних дослідженнях за участю 5813 дорослих пацієнтів з ожирінням або надмірною масою тіла із хоча б одним супутнім захворюванням, пов’язаним з надмірною масою тіла. Загалом найбільш частими побічними реакціями були розлади травної системи (67,9%) (див. розділ «Опис окремих побічних реакцій»).
Список побічних реакцій
Нижче зазначені побічні реакції, про які повідомлялося у дорослих. Побічні реакції класифіковані за системами органів та частотою виникнення. Оцінку частоти виникнення побічних реакцій проводили за такою шкалою: дуже часто (≥ 1/10), часто (від ≥ 1/100 до < 1/10), нечасто (від ≥ 1/1000 до < 1/100), рідко (від ≥ 1/10000 до < 1/1000), дуже рідко (< 1/10000) та частота невідома (не можна визначити на основі наявних даних). У кожній групі побічні реакції наведено в порядку зниження їхньої серйозності.
З боку імунної системи: рідко — анафілактичні реакції.
Порушення метаболізму і харчування: часто — гіпоглікемія*; нечасто — зневоднення.
Психічні розлади: часто — безсоння**.
З боку нервової системи: дуже часто — головний біль; часто — запаморочення, дисгевзія.
З боку серцево-судинної системи: нечасто — тахікардія.
З боку травної системи: дуже часто — нудота, блювання, діарея, запор; часто — сухість у роті, диспепсія, гастрит, гастроезофагеальна рефлюксна хвороба, біль у верхньому відділі черевної порожнини, метеоризм, еруктація, здуття живота; нечасто — панкреатит***, затримка випорожнення шлунка****; частота невідома — кишкова непрохідність†.
З боку печінки та жовчних шляхів: часто — жовчнокам’яна хвороба***; нечасто — холецистит***.
З боку шкіри та підшкірних тканин: часто — висип, нечасто — кропив’янка, невідомо — амілоїдоз шкіри.
З боку нирок та сечовивідних шляхів: рідко — гостра ниркова недостатність, порушення функції нирок.
Загальні розлади та реакції в місці ін’єкції: часто — реакції в місцях ін’єкцій, астенія, втома; нечасто — нездужання.
Лабораторні дослідження: часто – підвищений рівень ліпази, підвищений рівень амілази.
* Гіпоглікемія (базується на симптомах, про які повідомляли самі пацієнти і які не підтверджені вимірюваннями рівня глюкози в крові) виникала у пацієнтів, які не хворіють на цукровий діабет 2 типу та які застосовували лікарський засіб Саксенда® у поєднанні з дієтою та фізичною активністю. Для отримання додаткової інформації див. розділ «Опис окремих побічних реакцій».
** Безсоння в основному спостерігалося протягом перших 3 місяців лікування.
*** Див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій».
**** З контрольованої фази 2, 3а та 3б клінічних досліджень.
† Побічні реакції, відомі з постмаркетингового досвіду.
Опис окремих побічних реакцій
Гіпоглікемія в пацієнтів без цукрового діабету 2 типу
У процесі клінічних випробувань не було зафіксованого жодного тяжкого випадку виникнення гіпоглікемії (що потребує сторонньої допомоги) у пацієнтів з надмірною масою тіла чи ожирінням без цукрового діабету 2 типу, які застосовували лікарський засіб Саксенда® у поєднанні з дієтою та фізичною активністю. Про виникнення симптомів гіпоглікемії повідомили 1,6% пацієнтів, які отримували лікарський засіб Саксенда®, та 1,1% пацієнтів, які отримували плацебо. Однак ці випадки не були підтверджені вимірюванням рівня глюкози в крові. Більшість випадків легко переносились.
Гіпоглікемія у хворих на цукровий діабет 2 типу
У клінічному досліджені були повідомлення про випадки тяжкої гіпоглікемії у 0,7% пацієнтів з надмірною масою тіла чи ожирінням, які хворіли на цукровий діабет 2 типу й застосовували лікарський засіб Саксенда® у поєднанні з дієтою та фізичною активністю та які застосовували сульфонілсечовину. Також зафіксовано виникнення симптомів гіпоглікемії у 43,6% пацієнтів, які застосовували лікарський засіб Саксенда®, та у 27,3% пацієнтів, які отримували плацебо. Серед пацієнтів, які не застосовували сульфонілсечовину, було зафіксовано виникнення симптомів гіпоглікемії (визначаються концентрацією глюкози у плазмі крові ≤ 3,9 ммоль/л) у 15,7% пацієнтів, які отримували лікарський засіб Саксенда®, та у 7,6% пацієнтів, які отримували плацебо.
Гіпоглікемія у хворих на цукровий діабет 2 типу, які отримують інсулін
У процесі клінічного дослідження повідомлялося про випадки тяжкої гіпоглікемії (що потребує сторонньої допомоги) у 1,5% пацієнтів з надмірною масою тіла чи ожирінням, які хворіли на цукровий діабет 2 типу та застосовували інсулін та ліраглутид в дозі 3,0 мг/добу у поєднанні з дієтою та фізичною активністю і застосуванням до 2 пероральних протидіабетичних препаратів. В цьому дослідженні було зафіксовано виникнення симптоматичної гіпоглікемії (визначена як рівень глюкози в плазмі крові ≤ 3,9 ммоль/л, що супроводжується симптомами) у 47,2% пацієнтів, які отримували ліраглутид у дозі 3,0 мг/добу, та у 51,8% пацієнтів, які отримували плацебо. У групі застосування сульфонілсечовини були зафіксовані повідомлення про виникнення симптомів гіпоглікемії у 60,9% пацієнтів, які отримували ліраглутид 3,0 мг/добу, та у 60,0% пацієнтів, які отримували плацебо.
Розлади травної системи
Більшість випадків розладів травної системи були легкого або помірного ступеня тяжкості та не призводили до припинення терапії. Зазвичай реакції виникали протягом перших тижнів лікування і послаблювались протягом декількох днів або тижнів при продовженні лікування.
У пацієнтів віком від 65 років при застосуванні лікарського засобу Саксенда® частіше спостерігатися порушення з боку травної системи.
У пацієнтів з порушеннями функції нирок легкого або помірного ступеня тяжкості (кліренс креатиніну ≥ 30 мл/хв) при застосуванні лікарського засобу Саксенда® можуть частіше виникати порушення з боку травної системи.
Гостра ниркова недостатність
Зафіксовано випадки гострої ниркової недостатності у пацієнтів, які застосовували агоністи рецепторів ГПП-1. Більшість зареєстрованих випадків були у пацієнтів, у яких спостерігалися нудота, блювання та діарея, що і призводило до втрати рідини (див. розділ «Особливості застосування»).
Алергічні реакції
Були повідомлення про кілька випадків виникнення анафілактичних реакцій, що супроводжувались такими симптомами, як гіпотензія, підвищене серцебиття, напади задухи та набряки після застосування ліраглутиду. Анафілактичні реакції можуть бути небезпечними для життя. Тому, якщо є підозра на виникнення анафілактичної реакції, слід припинити застосування ліраглутиду (див. розділ «Протипоказання»).
Реакції в місці ін’єкції
Повідомлялось про реакції в місці введення лікарського засобу Саксенда®. Ці реакції зазвичай були легкими, більшість із них зникали в процесі подальшого лікування.
Тахікардія
У процесі клінічних досліджень були повідомлення про випадки тахікардії у 0,6% пацієнтів, які отримували лікарський засіб Саксенда®, та у 0,1% пацієнтів, які отримували плацебо. Більшість випадків були легкого або помірного ступеня тяжкості. Випадки були поодинокими, більшість із них зникали в процесі подальшого лікування.
Амілоїдоз шкіри
Амілоїдоз шкіри може виникнути в місці ін’єкції (див. розділ «Спосіб застосування та дози»).
Діти
У ході клінічного дослідження за участю дітей віком від 12 років із ожирінням 125 пацієнтів приймали лікарський засіб Саксенда® впродовж 56 тижнів.
Загалом частота, тип і тяжкість небажаних реакцій у дітей віком ≥ 12 років з ожирінням були зіставними із такими у дорослих пацієнтів. Порівняно з дорослими пацієнтами у дітей віком від 12 років удвічі частіше спостерігалось блювання.
Відсоток пацієнтів, які повідомили принаймні про один епізод клінічно значущої гіпоглікемії, був вищим у групі застосування ліраглутиду (1,6%), ніж у групі плацебо (0,8%). Під час проведення дослідження не зареєстровано жодного епізоду тяжкої гіпоглікемії.
Повідомлення про небажані реакції та відсутність ефективності лікарського засобу
Повідомлення про побічні реакції після реєстрації лікарського засобу має важливе значення. Це дає змогу проводити моніторинг співвідношення користь/ризик при застосуванні цього лікарського засобу. Медичним та фармацевтичним працівникам, а також пацієнтам або їхнім законним представникам слід повідомляти про усі випадки підозрюваних побічних реакцій та відсутності ефективності лікарського засобу через Автоматизовану інформаційну систему з фармаконагляду за посиланням https://aisf.dec.gov.ua.
Термін придатності
30 місяців.
Дату завершення терміну придатності зазначено на маркуванні шприц-ручки та картонній упаковці після слів «Придатн. до».
Після першого застосування — 1 місяць.
Умови зберігання
Зберігати в недоступному для дітей місці.
Зберігати в холодильнику (2–8 °С) подалі від морозильної камери.
Після першого застосування зберігати при температурі нижче 30 °С або в холодильнику (2–8 °С). Не заморожувати.
Для запобігання дії світла зберігати шприц-ручку із закритим ковпачком.
Несумісність
Додавання до лікарського засобу Саксенда® будь-якої субстанції може призвести до деградації ліраглутиду. Оскільки дослідження сумісності не проводилися, цей лікарський засіб не можна застосовувати з іншими лікарськими засобами.
Упаковка
Попередньо заповнена багатодозова одноразова шприц-ручка, виготовлена з поліпропілену, поліацеталу, полікарбонату та акрилонітрилу бутадієну стиролу, містить картридж (скло типу I) разом із поршнем (бромбутил) та ламіновану гумову прокладку (бромбутил/поліізопрен).
Кожна шприц-ручка містить 3 мл розчину, що дає можливість ввести дози по 0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг та 3,0 мг.
Упаковка містить 1, 3 або 5 попередньо заповнених шприц-ручок в картонній коробці.
Категорія відпуску
За рецептом.
Заявник/Виробник
А/Т Ново Нордіск.
Місцезнаходження заявника/виробника та адреса місця провадження його діяльності
Ново Аллє
Багсваерд, 2880
Данія.